

Abstract
Oral administration of monoclonal antibodies (mAbs) may enable the localized treatment of infections or other conditions in the gastrointestinal tract (GI) as well as systemic diseases. As with the development of oral protein biotherapeutics, one of the most challenging tasks in antibody therapies is the loss of biological activity due to physical and chemical instabilities. New families of complexation hydrogels with pH-responsive properties have demonstrated to be excellent transmucosal delivery vehicles. This contribution focuses on the design and evaluation of hydrogel carriers that will minimize the degradation and maximize the in vivo activity of anti-TNF-α, a mAb used for the treatment of inflammatory bowel disease (IBD) in the GI tract and systemically for the treatment of rheumatoid arthritis. P(MAA-g-EG) and P(MAA-co-NVP) hydrogels systems were optimized to achieve adequate swelling behavior, which translated into improved protein loading and release at neutral pH simulating the small intestine conditions. Additionally, these hydrogel systems preserve antibody bioactivity upon release resulting in the systemic circulation of an antibody capable of effectively performing its biological function. The compatibility if these hydrogels for mAb bioactivity preservation and release makes them candidates for use as oral delivery systems for therapeutic antibodies.
INTRODUCTION
Protein-based drugs have emerged as one of the most promising classes of therapeutics.1 In 2011, the Biotechnology Industry Organization reported ~200 protein-based medicines on the market and ~400 in development.2 Specifically, antibodies have become a major class of protein-based therapeutic agents3 with about one-fifth of new drugs in clinical testing today being antibody therapeutics.4
Antibody therapies have been widely used to provide immunity against diseases or to help fight off infections.5,6 As an example, anti-TNF-α antibodies are currently used for the treatment of inflammatory bowel diseases (IBDs), an inflammatory disorder of the gastrointestinal (GI) tract, and rheumatoid arthritis (RA), an autoimmune disease that causes abnormal inflammation levels that damage joints and organs.6,7 Anti-TNF-α antibodies have demonstrated high effcacy in treating IBDs and RA; however, because they are commonly delivered by injection (i.e., intravenous, subcutaneous, or intramuscular) and neutralize TNF throughout the body, their use is associated with serious side effects, including reactivation of tuberculosis and a long-term risk of malignancy.8 Moreover, the common route of administration and the large doses required make antibody therapies expensive and hardly accessible for patients.
There is, therefore, a developing interest in designing dosage forms of antibody therapeutics, which could be administered orally for the treatment of infections and other local conditions in the GI tract but also for systemic conditions such as RA.8 Oral dosage forms of antibodies would have the advantages of reduced cost due to the simplicity of the administration. However, as with all the orally administered proteins and peptides, orally delivered antibodies are subject to denaturation at the acidic pH of the stomach as well as degradation by proteases present in the stomach, small intestine, and to a lesser extent, the colon.8,9 Exposure to these harsh conditions may result in changes in structural conformation of the antibodies, which could lead to loss of biological activity (e.g., neutralizing activity).
Previous work in our group has focused on developing platforms for the delivery of insulin and other protein-based therapeutics via the oral route, including the development of the complexation hydrogel system poly(methacrylic acid-grafted-poly(ethylene glycol)) or P(MAA-g-EG).10 These complexation hydrogel carriers are suitable candidates for the oral delivery of proteins and peptides because their network structure exhibits pH-dependent swelling and changes in the network pore structure due to the reversible formation/dissociation of interpolymer complexes,11 responding to pH changes in the GI tract and providing protection to the encapsulated biomolecules from protease degradation
To address the medical need of improved antibody therapies, we designed and evaluated orally deliverable anti-TNF-α antibody systems by optimizing the hydrogel-based carriers to minimize the degradation and maximize the in vivo activity of anti-TNF-α a mAb. Hydrogel systems were designed to optimize anti-TNF-α loading and release. A detailed evaluation on protein structure and in vitro bioactivity was performed to allow for the rational screening and selection of appropriate polymer composition. Finally, prescreened anti-TNF-α loaded hydrogels were tested in an ex vivo closed-loop model. The studies described herein demonstrate that the designed hydrogel vehicles preserved antibody bioactivity suggesting that this platform can be effectively used as a orally delivery system for therapeutic antibodies.
MATERIALS AND METHODS
Materials
Methacrylic acid (MAA), N-vinylpyrroidone (NVP), tetraethylene glycol dimethacrylate (TEGDMA), and 1-hydroxycyclohexyl phenyl ketone (Irgacure 184) were obtained from Sigma-Aldrich (St. Louis, MO). Poly(ethylene glycol) monomethyl ether monomethacrylate (PEGMMA, 1000 g mol−1), poly(ethylene glycol) 1000 dimethacrylate (PEG1000DMA, with nominal PEG molecular weight of 1000 g mol−1, corresponding to 23 repeating units) and poly(ethylene glycol) 400 dimethacrylate (PEG400DMA, with nominal PEG molecular weight of 400 g mol−1, corresponding to 9 repeating units) were purchased from Polysciences, Inc. (Warrington, PA). All chemicals were used as received except for MAA which was vacuum distilled at 54 °C and 25 mmHg prior to use to remove the inhibitor hydroquinone. All solvents were of ACS or HPLC grade.
Synthesis of P(MAA-g-EG) and P(MAA-co-NVP) Hydrogel Microparticles
P(MAA-g-EG) and P(MAA-co-NVP) hydrogel microparticles were synthesized with three different cross-linkers added to the monomer mixture at the same cross-linking density. The cross-linkers used in this work were TEGDMA, PEG400DMA, and PEG1000DMA.
Polymer films were prepared by UV-initiated free radical polymerization of MAA and PAGMMA or MAA and NVP.11 In the synthesis of these materials, the monomer content is defined as the monomers used to form the polymeric backbone and the cross-linking agent that yields the covalently bonded network. Monomers were mixed in the molar ratio of 1:1. The cross-linkers were added at a concentration of 0.75 mol % of the total monomer content. The photoinitiator Irgacure-184 was added in the amount of 0.1 wt % of the total amount of monomers. The monomers, cross-linking agent, and photoinitiator mixture was diluted in 50% (v/v) ethanol and briefly sonicated (Bransonic 8510, Branson Ultrasonic Corp., Danbury, CT) to homogeneity. The polymerization of hydrogel films was carried out under a nitrogen environment in a sealed glovebox by exposing the sonicated mixture contained between glass plates to UV light (Dymax 2000 Light Curing System, Torrington, CT) at an intensity of 16–17 mW cm−2 for 30 min. Films were then washed with deionized water and dried (≤24 h at RT and 48 h at 30 °C under vacuum). Dried films were crushed and sieved to ≤75 μm to obtain hydrogel microparticles. As has been reported from macroscopic imaging analysis of similar hydrogel systems designed in our group, the morphology of these hydrogel particles is irregular, as expected from particles created by crushing and sieving, and their surface contains a mixture of smooth and rough areas (data not shown).12,13
Hydrogel Swelling Characterization
Equilibrium swelling studies were used to study the pH-responsive behavior of P(MAA-g-EG and P(MAA-co-NVP) hydrogel films. A 9 mm (aspect ratio of 18) punch was used to cut discs from the polymer films after purification but prior to film drying. Hydrogel discs were dried at room temperature for about a day and then vacuum-dried for 48 h at 30 °C. Equilibrium swelling behavior was determined by swelling the hydrogel discs in buffer solutions at 37 °C for 24 h. For the pH range between 3.2 and 8.0 the discs were placed in β,β-dimethylglutaric acid (DMGA) buffer, which maintained ionic strength using 0.1 N sodium chloride (NaCl), whereas samples at a pH of 2.0 were placed in pepsin-free simulated gastric fluid (0.1 N HCl, 2 g/L NaCl). After the incubation period, the swollen hydrogels were carefully blotted to remove excess solution, and weighted to determine equilibrium weight swelling ratio (q).
In Vitro Microparticle Cytotoxicity
L929 fibroblasts cell line (American Type Culture Collection, ATCC, Rockwell, MD) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin and streptomycin, at 37 °C in humidified environment of 5% CO2 (Sheldon Signature HEPA Clean CO2 incubator, VWR). For cytotoxicity experiments, cells were detached mechanically by carefully scrapping the T-75 flask surface and adjusted to the required concentration of viable cells, by counting in a hemocytometer.
L929 fibroblasts cells were seeded at 20 000 cells per well in 96 well-plates (Thermo Scientific, Waltham, MA) in phenol red-free DMEM with 2% FBS. Tissue culture plates containing cells were incubated at 37 °C, 5% CO2 for 24 h before testing. On the testing day, cells were incubated with particle suspensions at concentrations of 1, 0.5, 0.25, and 0.125 mg/mL in phenol red-free DMEM with 2% FBS. Live control wells (medium only) and lysed control wells were included. Following 2 and 48 h of incubation, the particles were removed and the cells were rinsed 3× with sterile Hank’s balanced salt solution (HBSS). Cell viability was then assayed by adding the CellTiter Aqueous One Solution reagent (MTS, Promega) directly to the culture wells and incubating for 2 h. Finally, the absorbance at 690 nm (background) and 490 nm (MTS assay) was recorded using a microplate reader (Synergy HT, BioTek Instruments, Inc., Winooski, VT). The 690 nm readings from each well were subtracted from absorbance (OD) values at 490 nm, and the viability fraction was defined as
| (1) |
Anti-TNF-α Antibody Loading into Hydrogel Microparticles
Anti-TNF-α antibody (clone XT3.11, BioXCell Fermentation/Purification Services, West Lebanon, NH) was loaded into hydrogel microparticles by equilibrium partitioning.14 Initially, microparticles were incubated overnight at a concentration of 10 mg/mL in 10 mM phosphate buffer (PBS, pH 7.4) in low-protein binging centrifuge tubes with constant end-to-end rotation to ensure wetting and swelling of the hydrogel microparticles. An initial sample of the particle suspension supernatant was taken and replaced with fresh buffer. Anti-TNF-α antibody stock solution (2 mg/mL in PBS, pH 7.4) was transferred to microparticles suspension to achieve a protein concentration of 1 mg/mL and incubated at room temperature (RT) with constant end-to-end rotation. After 12 h of incubation, the particles were collapsed by the addition of 0.1 N HCl until reaching a pH 3.5. The supernatant was removed and its pH was adjusted to 7.4 with 0.1 N NaOH, and stored at 4 °C until assayed. Microparticles were rinsed with water and 0.1 N HCl to remove any surface-absorbed protein. Finally, Anti-TNF-α antibody-loaded microparticles were freeze-dried.
Determination of anti-TNF-α antibody concentrations in supernatants was carried out with a BCA Protein Assay Kit (Thermo Scientific, Sugar Land, TX). The apparent loading effciency was calculated on the basis of the anti-TNF-α antibody concentration in the supernatant before and after loading.
In Vitro Anti-TNF-α Antibody Release from Hydrogel Carrier under Dynamic Conditions
Release studies aimed to simulate the pH changes the hydrogel microparticles would experience in vivo when passing through the stomach and into the small intestine (i.e., from an acidic pH to a neutral pH).14 For this experiment, 10 mg of anti-TNF-α antibody-loaded microparticles were suspended in DMGA buffer at pH 3.2 with an ionic strength of 0.1 M at a concentration of 1 mg/mL. Samples of 200 μL were withdrawn at predetermined time intervals up to 90 min and replaced with fresh buffer at pH 3.2 while microparticles suspensions were maintained at 37 °C with constant end-to-end rotation. After 90 min, pH was titrated to 7.4 by careful addition of 1 N NaOH. Samples were taken and replaced with fresh buffer at predetermined time intervals up to 3 h. The determination of anti-TNF-α antibody concentration in the supernatant samples was carried out with a Micro BCA Protein Assay Kit (Thermo Scientific, Sugar Land, TX).
Antibody Structural Stability after Release
The structural stability of anti-TNF-α after release from hydrogel microparticles was assessed by circular dichroism and fluorescence spectroscopy as described elsewhere.15,16 UV circular dichroism (CD) was used to evaluate protein secondary structure using a Jasco J-710 spectropolarimeter (Jasco, Easton, MD) at a wavelength range of 190–260 nm.15,17–19 For tertiary structure protein analysis, a Cary 5000 spectrometer (Varian, Inc., Australia) was used to collect fluorescence spectra (300–500 nm) at an excitation wavelength of 280 nm that corresponds to tyrosine and tryptophan residues.18,19
Collected CD and fluorescence spectra for released and unencapsulated antibody’s were analyzed for peak intensity and location (wavelength) shifts compared to that of uncapsulated antibody control, which are indicative of alterations in protein structure.18,19 Representative data out of three independent analyses is presented.
Anti-TNF-α Residual Functionality after Release
Functionality of anti-TNF-α antibody is described as the antibody ability to recognize and bind to TNF-α and was assessed via enzyme-linked immunosorbent assay (ELISA). Briefly, high protein binding 96-well Costar microtiter plates (Corning Life Sciences, Lowell, MA) were coated overnight with recombinant murine TNF-α (PeproTech, Rocky Hill, NJ) in PBS at a concentration of 5 μg/mL. After blocking for 2 h at RT with 3% (w/v) bovine serum albumin (BSA, Sigma-Aldrich) in PBS-T (PBS containing 0.05% (v/v) Tween 20), the released antibody samples as well as unencapsulated anti-TNF-α were added to their respective wells at a concentration of 10 μg/mL and incubated for 90 min at 4 °C. Alkaline phosphatase-conjugated Goat Anti-Rat IgG was added as detection antibody and the colorimetric reaction was developed at RT for 50 min using phosphatase substrate. The optical density (OD) of each well was measured at 405 nm using a microplate reader (Synergy HT, BioTek Instruments, Inc., Winooski, VT). Final results are presented as residual functionality, which represent the normalization of the data by the unencapsulated antibody control.
In Vitro Anti-TNF-α Residual Bioactivity after Release from Hydrogel Microparticles
An in vitro potency assay based on the TNF-α-induced cytotoxicity on L929 murine fibroblast cells20,21 was utilized to determine the maintenance of antibody functionality after release from hydrogel microparticles. For this assay, L929 fibroblast were cultured and harvested as described in the In Vitro Microparticle Cytotoxicity section. After being harvested, cells were seeded into 96-well flat-bottomed plates at a concentration of 3.5 × 104 cells per well and incubated overnight at 37 °C in 5% CO2.
Samples of antibody released from different formulations of hydrogel microparticles and uncapsulated antibody control were diluted at a uniform concentration (85% effective dose (ED)85 determined for the uncapsulated control) and preincubated with a fixed amount of TNF-α (250 pg/mL) for 4 h at 37 °C. Samples were then transferred to corresponding wells on L929 culture plates and incubated for additional 24 h in the presence of actinomycin D at 37 °C.20,21 Cells were finally assayed for cell viability using the CellTiter Aqueous One Solution reagent (MTS, Promega) directly to the culture as previously described. Residual in vitro bioactivity was determined by normalizing OD values from each sample to the values obtained by cells cultured in the absence of TNF-α.
Ex Vivo Closed-Loop Studies To Evaluate Antibody Bioactivity
Animals
Sprague-Dawley rats with jugular vein catheters were purchased from Taconic (Hudson, NY). All rats were housed under specific pathogen-free conditions where all bedding, caging, and feed were sterilized prior to use. All animal procedures were conducted with the approval of the Temple University Institutional Animal Care and Use Committee.
Adult rats entered the Temple animal facility, catheters were flushed and maintained as recommended by the manufacturer, and before surgery, a liquid diet (LD101 formulation, Test Diet, St. Louis, MO) was implemented for at least 72 h to help clear the intestinal line of obstructions. The rats were then fasted overnight (<24 h) prior to surgery for which only water was made available.
Closed-Loop Surgical Procedure and Treatments
An in situ closed-loop method was employed to investigate the bioavailability of anti-TNF-α antibody following direct administration of antibody-loaded hydrogel microparticle formulations to intestinal segments of rats.22 On the day of surgery, rats were placed under anesthesia with isoflurane (induction dose of 5% in oxygen followed by 1.5%–3% during procedure) and placed on heating pads to maintain the proper body temperature. Once the surgical plane of anesthesia had been reached, a 6 cm abdominal midline incision was made, exposing the organs and abdominal cavity, and giving access to the intestines. An approximate 10 cm section of the ileum was isolated and tied off carefully using suture thread to ensure that blood circulation is not compromised creating a closed loop. The intestine was then placed back into the cavity, and the wound temporarily stapled shut. Rats were then left to rest for at least 30 min to recover from surgical trauma. Afterward, a blood sample was taken to establish baseline plasma levels.
Then, the intestinal incision was reopened and hydrogel formulations were directly injected using a 1-in., 25-gauge needle inside the intestinal loop. The hydrogel formulations included in this close-loop study were anti-TNF-α-loaded P(MAA-g-EG) and anti-TNF-α-loaded P(MAA-co-NVP) hydrogel microparticles. The dose of anti-TNF-α loaded into hydrogel microparticles was 70 μg/kg body weight.
The isolated intestinal section was then placed back within the abdominal cavity, and the incision was closed again with staples. Blood samples were then taken at 5, 10, 15, 30, 60, 120, 180, and 240 min from the jugular vein using the preadapted catheters. After every blood sample was taken, catheters were flushed as recommended by manufacturer to avoid clogging and reduce chances for sample contamination. Once the last blood sample was been taken, the rats were euthanized via an overdose of isoflurane (5% in oxygen), with a bilateral thoracotomy performed after breathing had ceased as a second method of euthanasia.
Antibody Detection in Serum and Functionality
Anti-TNF-α antibody in serum was measured by ELISA as previously described. Semiquantitative levels of anti-TNF-α were calculated from a standard of anti-TNF-α antibody that was included in each assay.
In vitro potency L929 bioassay described in the previous section of this manuscript was utilized to verify the neutralization activity of the released anti-TNF-α mAb.
Statistical Analysis
Statistical ANOVA analysis was carried out using JMP 7 software (SAS Institute, Cary, NC). Comparisons for the multiple formulations were achieved using Turkey’s HSD analysis with differences considered significant when p ≤ 0.05.
RESULTS AND DISCUSSION
Higher Swelling Ratio Was Obtained with P(MAA-g-EG) Hydrogels and Those Formulations Containing More Hydrophilic Cross-Linkers
Dynamic swelling experiments were completed to study the pH-responsive behavior of the polymer protein carriers in simulated physiological fluids and the effects of the various cross-linkers utilized for hydrogel synthesis. In this type of pH-responsive systems, the polymer network contains weak acid or base groups that undergo complexation by hydrogen bonding, thus exhibiting pH-dependent swelling properties with fast expansion and contraction.23 In the case of acrylic acid-based copolymers, the monomer MAA provides the weak acid groups that allow for swelling performance.23,24 As shown in Figure 1, minimal swelling occurred in gastric conditions (i.e., low pH) due to the anionic nature of these systems and the hydrogen bonding between the carboxyl groups on the MAA moiety and the etheric oxygen of the PEG chain. Once the pKa (4.8–4.9) of these carboxyl groups is exceeded, swelling of the system is observed.25,26
Figure 1.

Higher swelling ratio was obtained with P(MAA-g-EG) hydrogels and those formulations containing more hydrophilic cross-linkers. pH-dependent equilibrium weight swelling ratio, q, of (A) P(MAA-g-EG) and (B) P(MAA-co-NVP) hydrogel disks synthesized with TEGDMA, PEGDMA-400, and PEGDMA-1000 as cross-linkers. Disks from each formulation were placed in DMGA buffer (pH value ranging from 2.0 to 8.0) for 15 min. The weight–swelling ratio was calculated by dividing the swollen weight of the disk by the dry weight of the disk.
When the equilibrium swelling ratio of the two hydrogel systems tested in this study is compared, a higher swelling ratio is observed for P(MAA-g-EG) hydrogels (Figure 1). The lower swelling weight ratio of P(MAA-co-NVP) may be the result of an improved hydrogen-bonding strength between MAA and NVP.27,28 In both hydrogel systems, a slight increase in hydrogel swelling weight ratio was observed as the hydrophilicity of the incorporated cross-linker increases (from TEGDMA to PEGDMA-1000). The PEGDMA-1000 has approximately 23 ethylene glycol units where as PEGDMA-400 and TEGDMA have ~9 and 4 ethylene glycol units, respectively. Incorporating cross-linkers with higher hydrophilicity may help the network swell to a greater extent in the aqueous medium. Thus, by changing the nature of the cross-linker used to synthesize these hydrogel systems, the hydrogel network structure could be optimized (i.e., increase in the mesh size)13,29 for the delivery of large molecular weight proteins such as immunoglobulins.
In Vitro Biocompatibility of Hydrogel Microparticles Was Demonstrated in Fibroblast (L929) Cell Cultures
Fibroblast (L929) cells were used to determine biocompatibility of P(MAA-g-EG) and P(MAA-co-NVP) microparticles. Several parameters such as hydrogel composition, cross-linker type, microparticles size, and microparticle concentration were evaluated. Results and observations are summarized in Table 1. Cell viability was determined using MTS assay after 2 and 48 h of incubation with the microparticles. Similar responses were observed when cells were exposed to the different carriers formulations, showing low or absence of cytotoxic effect. Only when microparticles of size higher than 75 μm or particle concentrations equal or higher than 0.5 mg/mL were tested was a slight decrease in cell viability observed (data not shown).
Table 1.
Summary of Cytotoxicity Response Observed after P(MAA-g-EG) and P(MAA-co-NVP) Microparticles Were Incubated in L929 Fibroblast Cell Cultures for 24 h
| parameter evaluated | cytotoxicity response in L929 fibroblast cell cultures |
|---|---|
| hydrogel chemistry | maintain low cytotoxicity |
| cross-linker | maintain low cytotoxicity |
| increase in microgel size | increase cytotoxicity |
| increase in microgel concentration |
increase cytotoxicity |
Hydrogel-Swelling Behavior Translated into Improved Protein Loading and Release Capabilities
Previous work within our group has demonstrated the effcacy of the tested hydrogel systems to encapsulate several proteins (e.g., insulin, calcitonin,22,30,31 and growth hormone). However, all the proteins tested until now have relative low molecular weight compared to immunoglobulin, which average molecular weight is 150 kDa and have a more define globular conformation. Anti-TNF-α antibodies were successfully loaded into both P(MAA-g-EG) and P(MAA-co-NVP) hydrogel microparticles (Figure 2A). However, while common loading percentages of 90% were observed for proteins such as insulin,22,31 the observed loading effciencies for anti-TNF-α antibodies were between 40 and 60%. Results from Figure 2A show that for P(MAA-g-EG)-based formulations, the loading effciencies were above 50% while for P(MAA-co-NVP)-based systems loading effciencies ≤50% were obtained. The observations of different loading profiles between P(MAA-g-EG) and P(MAA-co-NVP)-based systems is a result of the different equilibrium swelling behavior observed for both chemistries as previously discussed (Figure 1). The apparent loading effciencies of the antibody were not affected by the size of the cross-linking molecule, as no statistical significance was obtained between the formulations with different cross-linkers.
Figure 2.

Hydrogel-swelling behavior translated into improved protein loading and release capacities. (A) Loading effciency of anti-TNF-α antibody in P(MAA-g-EG) and P(MAA-co-NVP) microparticles (≤75 μm) formulated with TEGDMA, PEGDMA-400, and PEGDMA-1000 as cross-linkers. Hydrogel microparticles were incubated in an anti-TNF-α solution in PBS (pH 7.4), and 1 N HCl was added to the solution to lower the pH and collapse the microparticles. (B) Anti-TNF-α antibody release from P(MAA-g-EG) hydrogel microparticles synthesized with different cross-linkers: PEGDMA-1000, PEGDMA-400, and TEGDMA. Protein release from the microparticles was studied under dynamic pH conditions (from pH 3.0 to pH 7.4) to mimic the transit of the particles through the GI tract. Released protein was quantified using MicroBCA protein assay.
In vitro release studies were performed at a two-step pH change. To mimic the transition of the polymer formulations from the gastric environment to the upper small intestine, the pH of the release buffer was changed from pH 3.2 to 7.4 after 90 min of initial incubation at pH 3.2. Figure 2B shows the release profile of P(MAA-g-EG) microparticles prepared with the different cross-linkers. At low pH conditions, low amounts of released anti-TNF-α antibody are detected, reaching a maximum of 10–50% at this pH depending on the specific cross-linking agent. Microparticles prepared with PEGDMA-1000, the more hydrophilic cross-linking, showed the higher amount of antibody released at low pH conditions. This observation may be a result of a higher mesh size of the network at the gastric pH for formulations containing the longer cross-linker PEGDMA-1000, which allow protein leaking at low pH.
After 90 min, the pH was stepped from 3.2 to 7.4 and an immediate increase in the amount of antibody released was observed for all polymer carriers and reached a maximum in about 2 h (Figure 2B). Microparticles prepared with PEGDMA-1000 cross-linker exhibited a faster release rate than PEGDMA-400 or TEGDMA-containing hydrogels. This can be explained by the presence of protein at or near the surface of the polymer as a result of the protein diffusion already initiated in low pH conditions. Similar released profiles were observed for P(MAA-co-NVP) microparticles prepared with the different cross-linkers; however, less amount of antibody was released from P(MAA-co-NVP) microparticles at low pH (data not shown). This is consistent with the equilibrium swelling profile observed for these formulations corroborating the stronger hydrogen bonding between MAA and NVP.30
On the basis of these loading and release results, TEGDMA was chosen as the cross-linker to move forward for in vitro functionality and stability studies as well as ex vivo studies. More hydrophilic cross-linker, longer cross-linker agents (i.e., PEGDMA-400 and PEGDMA-1000), showed higher percentages of protein released at low pH, which could translate to higher protein losses while the carriers are passing through the gastric environment of the stomach and therefore a lower amount of antibody will reach the site of action or absorption (i.e., intestinal area) in vivo.
Anti-TNF-α Antibody in Vitro Neutralization Activity Is Maintained upon Release from P(MAA-g-EG) Microparticles
An in vitro TNF-α neutralization assay was employed to assess the ability of anti-TNF-α-loaded hydrogel microparticles to preserve the biological function of the monoclonal antibody after loading and release from these carriers. Initial studies were performed in order to determine the optimal TNF-α cytotoxic dose and anti-TNF-α antibody neutralization dose to use in the assay. From these initial studies, it was determined that a 500 pg/mL dose of TNF-α was suffcient to achieve around 95% of cytotoxicity in L929 cells, while a dose of 5 μg/mL of anti-TNF-α antibody was necessary to protect close to 100% of cells (Figure 3A).
Figure 3.

Anti-TNF-α antibody in vitro neutralization activity is maintained upon release from P(MAA-g-EG) microparticles. Optimization of TNF-α cytotoxic dose and anti-TNF-α antibody neutralization dose. A 500 pg/mL dose of TNF-α was suffcient to achieve around 95% of cytotoxicity in L929 cells, while 5 μg/mL of anti-TNF-α antibody was necessary to protect close to 100% of cells. (A) Titration of cytolytic activity of the recombinant TNF-α. (B) Neutralization of TNF-α by different doses of antibod. The viability of the cells was quantified by MTS assay and represented as percentages against control cells cultured in the absence of TNF-α. Error bars represents standard deviation of three independent experiments performed in triplicate. (C) Anti-TNF-α-loaded P(MAA-g-EG) microparticles are capable of releasing bioactive antibody, while loading and release from P(MAA-co-NVP) microparticles affects antibody bioactivity. Percentage residual bioactivity from TNF-α cytolytic activity achieved by anti-TNF-α antibody. L929 cells were cultured for 24 h in the presence of TNF-α and unencapsulated antibody, or antibodies released from microparticles. The viability of the cells was quantified by the MTS assay and represented as percentages against control cells cultured in the absence of TNF-α. Error bars indicate standard error of three independent experiments performed in triplicate.
To test the in vitro bioactivity of antibody released from hydrogel microparticles, anti-TNF-α antibody was released from P(MAA-g-EG) and P(MAA-co-NVP) microparticles at pH 7.4 for at least 6 h at 37 °C. After release period was completed, supernatants containing released antibody were collected and concentrated to be able to employ a uniform concentration (5 μg/mL) for all tested formulations in the assay well. Unencapsulated antibody incubated for ~6 h at 37 °C in PBS, pH 7.4, was used as control and was also added at a concentration of 5 μg/mL to allow for direct comparison between the different groups.
As observed in Figure 3B, which showed residual bioactivity results for anti-TNF-α antibody released from P(MAA-g-EG) and P(MAA-co-NVP) microparticles synthesized using TEGDMA cross-linker, the bioactivity of this monoclonal antibody was preserved, as evidence by the protection of L929 cells from the cytotoxic effect of recombinant TNF-α, when released from P(MAA-g-EG) microparticles with protection levels similar or higher than ~90%. Anti-TNF-α antibody released from P(MAA-co-NVP) microparticles showed a 40% loss of bioactivity (Figure 3B), which suggests that specific interactions between the NVP monomer and the protein are occurring causing a decrease in the ability of the antibody to neutralize its target molecule (i.e., TNF-α). Residual antibody bioactivity results were consistent with residual antibody functionality results as evaluated by ELISA (data not shown).
Structural Integrity of Anti-TNF-α Antibody after Release from Hydrogel Microparticles
To get some insights on any structural changes that may be occurring to the anti-TNF-α mAb during loading and release from P(MAA-co-NVP) and that resulted in the observed bioactivity losses, a detailed structural analysis on protein released from hydrogel microparticles was performed. In specific, changes in the secondary and tertiary structure of the protein were evaluated and compared with uncapsulated protein control.
It has been previously reported that immunoglobulins have a predominance of β-sheet conformation, which is characterized by a single dominant minimum between 217 and 219 nm in CD spectra.32 This characteristic CD spectrum was observed for the anti-TNF-α mAb uncapsulated control with a minimum around the 219 nm, as shown in Figure 4A. However, shifts in the position of the characteristic minimum as well as differences in molar ellipticity (intensity) were observed for antibodies released from the tested hydrogel microparticle formulations. While antibodies released from P(MAA-g-EG) showed a slight shift of the minimum to the left (from 219 nm to ~216 nm) compared to that of the unencapsulated control, loss in the secondary structure of the antibody released from P(MAA-co-NVP) hydrogels is evidenced by the pronounced shift of the characteristic minimum as well as a significant increase in molar ellipticity (Figure 4A).
Figure 4.

Structural integrity of anti-TNF-α antibody after release from hydrogel microparticles. Representative data from three independent experiments is presented: (A) secondary structure by CD spectra and (B) tertiary structure by fluorescence spectroscopy.
For tertiary structure analysis, both maximum peak position and peak intensity changes in the fluorescence spectra are related to folding changes in the protein.17,18 For the anti-TNF-α antibodies released from both hydrogel chemistries, changes in the maximum position were not observed; however, a notorious decrease in the peak intensity was observed in the spectra of the antibody released from both microparticle formulations suggesting a potential unfolding of the antibody (Figure 4B). The intensity decrease was higher for antibodies released from P(MAA-co-NVP) microparticles, which is a result of the change in the availability of tryptophan and tyrosine residues on the protein surface due to a change in conformation.18
Bioavailability and ex Vivo Bioactivity of Anti-TNF-α Antibody upon Release from Hydrogel Microparticles
In the present study, an in situ closed-loop method was employed to investigate the bioavailability and ex vivo functionality of anti-TNF-α antibody released from hydrogel microparticles following direct administration to an intestinal section. The antibody absorption profile and ex vivo bioactivity characterization upon the intestinal administration of hydrogel formulation to rats at a dose of 70 μg/kg body weight are shown in Figure 5. Significant levels of anti-TNF-α antibody were detected in serum for both P(MAA-g-EG) and P(MAA-co-NVP) microparticles; suggesting that antibody is being released from the complexation hydrogels and absorbed through the intestinal mucosa. Maximum levels of antibody in serum were observed ~15 min after injection in the intestinal section and antibody serum levels between 3 and 6 μg/mL were still detected after 4 h of injection. When both hydrogel formulations were compared, higher antibody bioavailability was observed with the P(MAA-co-NVP) formulation. A control group consisting of soluble anti-TNF-α antibody delivered by subcutaneous administration was used in this study (data not shown) in order to compare the effcacy of our hydrogel systems to current antibody therapies. Rats that received subcutaneous administration of soluble antibody showed significant levels of antibody in serum during the first 60 min after administration but rapidly declined after that time until antibody levels were not identified by the analytical assay utilized in this study (data not shown).
Figure 5.
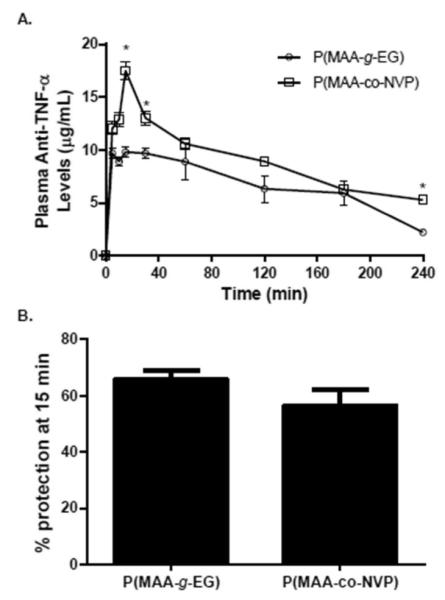
Bioavailability and ex-vivo bioactivity of anti-TNF-α antibody upon release from hydrogel microparticles. (A) Plasma anti-TNF-α levels versus time profiles following direct injection of anti-TNF-α-loaded P(MAA-g-EG) microparticles (n = 6) and anti-TNF-α-loaded P(MAA-co-NVP) microparticles (n = 6) into an intestinal closed-loop in healthy adult Sprague-Dawley rats. Blood samples were then taken at 5, 10, 15, 30, 60, 120, 180, and 240 min. The dose of anti-TNF-α loaded into microparticles was 70 μg/kg body weight. Anti-TNF-α mAb concentration in diluted serum was measured by ELISA (B) L929 cell TNF-α neutralization bioassay was utilized to assess the functionality of the released anti-TNF-α antibody in serum taken 15 min after hydrogel formulation injection into the intestinal loop. Concentration and % protection are presented as mean ± SEM The asterisk (*) represents a statistically significant difference from all other treatments at p ≤ 0.05.
Figure 5B demonstrates that both microparticle formulations deliver anti-TNF-α antibodies with equivalent TNF-α neutralization ability. The equivalent bioactivity demonstrated by anti-TNF-α released from P(MAA-g-EG) microparticles occurred while lower concentrations of antibodies were maintained in circulation compared to antibodies released from P(MAA-co-NVP) microparticles (Figure 5A). This corroborates the in vitro observations demonstrating the ability of P(MAA-g-EG) microparticles to provide an adequate stabilization and release functional anti-TNF-α mAb.
The observed absorption of functional anti-TNF-α antibodies through the intestinal mucosa into circulation after release from P(MAA-g-EG) and P(MAA-co-NVP) may translate into effective formulations for the treatment of diseases such as rheumatoid arthritis in which the neutralization of excess levels of TNF-α in circulation is required. However, considering that for most protein formulations delivered orally low bioavailability percentages in serum have been observed,10 it is most likely that not all the antibodies released from the hydrogel microparticles in the small intestine were transported into circulation, which opens the possibility to explore the use of these hydrogel formulations for the localized treatment of inflammatory bowel diseases.7
CONCLUSION
Some of the most diffcult challenges to improve current antibody-based therapies is the design of effective formulations that allow for the use of more patient-friendly administrations routes (i.e., oral administration) avoiding the side effects of large antibody doses administered intravenously. The studies reported herein demonstrated the potential use of complexation hydrogel systems (i.e., P(MAA-g-EG) and P(MAA-co-NVP microparticles) as an effective antibody oral delivery platform. These hydrogel microparticles provide the ability to protect the antibody from the harsh acidic environment of the GI tract and release it in the neutral pH of the small intestine where the antibody can be transported to systemic circulation. Additionally, these particles preserve antibody bioactivity upon release resulting in the systemic circulation of antibodies capable of effectively performing their biological function (i.e., neutralization of TNF-α cytokine). Complementary studies are required to investigate the effcacy of the formulations based on these particles for in vivo protection in a disease model.
ACKNOWLEDGMENTS
We thank the Center of Nano- and Molecular Science at The University of Texas at Austin for facilitating the use of CD spectrophotometer and fluorimeter.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Biotechnology in Health Care . Editors’ and Reporters’ Guide to Biotechnology. Biotechnology Industry Organization; 2011. [Google Scholar]
- (2).Healing the World . Editors’ and Reporters’ Guide to Biotechnology. Biotechnology Industry Organization; 2011. [Google Scholar]
- (3).Grainger DW. Controlled-release and local delivery of therapeutic antibodies. Expert Opin. Biol. Ther. 2004;4:1029–1044. doi: 10.1517/14712598.4.7.1029. [DOI] [PubMed] [Google Scholar]
- (4).Presta LG. Engineering antibodies for therapy. Curr. Pharm. Biotechnol. 2002;3:237–256. doi: 10.2174/1389201023378256. [DOI] [PubMed] [Google Scholar]
- (5).Casadevall A. Passive antibody therapies: Progress and continuing challenges. Clin. Immunol. 1999;93:5–15. doi: 10.1006/clim.1999.4768. [DOI] [PubMed] [Google Scholar]
- (6).Casadevall A, Dadachova E, Pirofski L. Passive antibody therapy for infectious diseases. Nat. Rev. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- (7).Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discovery. 2003;2:52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- (8).Reilly RM, Domingo R, Sandhu J. Oral delivery of antibodies: Future pharmacokinetic trends. Clin. Pharmacokin. 1997;32:313–323. doi: 10.2165/00003088-199732040-00004. [DOI] [PubMed] [Google Scholar]
- (9).des Rieuxa A, Fieveza V, Garinota M, Schneiderb Y-J, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Controlled Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- (10).Morishitaa M, Gotoa T, Nakamuraa K, Lowman AM, Takayamaa K, Peppas NA. Novel oral insulin delivery systems based on complexation polymer hydrogels: Single and multiple administration studies in type 1 and 2 diabetic rats. J. Controlled Release. 2006;110:587–594. doi: 10.1016/j.jconrel.2005.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lowman AM, Morishita M, Kajita M, Nagai T, Peppas NA. Oral delivery of insulin using pH-responsive complexation gels. J. Pharm. Sci. 1999;88:933–937. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- (12).Betancourt T, Pardo J, Soo K, Peppas NA. Characterization of pH-responsive hydrogels of poly(itaconic acid-g-ethylene glycol) prepared by UV-initiated free radical polymerization as biomaterials for oral delivery of bioactive agents. J. Biomed. Mater. Res., Part A. 2010;93:175–188. doi: 10.1002/jbm.a.32510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Carr DA, Peppas NA. Molecular structure of physiologically-responsive hydrogels controls diffusive behavior. Macromol. Biosci. 2009;9:497–505. doi: 10.1002/mabi.200800235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wood KM, Stone GM, Peppas NA. Wheat germ agglutinin functionalized complexation hydrogels for oral insulin delivery. Biomacromolecules. 2008;9:1293–1298. doi: 10.1021/bm701274p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Carrillo-Conde B, Schiltz E, Yu J, Phillips G, Wannemuehler MJ, Narasimhan B, Minion C. Encapsulation into amphiphilic polyanhydride microparticles stabilizes Yersinia pestis antigens. Acta Biomater. 2010;6:3110–3119. doi: 10.1016/j.actbio.2010.01.040. [DOI] [PubMed] [Google Scholar]
- (16).Carrillo-Conde BR, Darling RJ, Seilera SJ, Ramer-Taitb AE, Wannemuehler MJ, Narasimhan B. Sustained release and stabilization of therapeutic antibodies using amphiphilic polyanhydride nanoparticles. Chem. Eng. Sci. 2015;125:98–107. [Google Scholar]
- (17).Determan AS, Lin VSY, Nilsen-Hamilton M, Narasimhan B. Encapsulation, stabilization, and release of BSA-FITC from polyanhydride microspheres. J. Controlled Release. 2004;100:97–109. doi: 10.1016/j.jconrel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- (18).Determan AS, Wilson JH, Kipper MJ, Wannemuehler MJ, Narasimhan B. Protein stability in the presence of polymer degradation products: Consequences for controlled release formulations. Biomaterials. 2006;27:3312–3320. doi: 10.1016/j.biomaterials.2006.01.054. [DOI] [PubMed] [Google Scholar]
- (19).Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials. 2006;28:108–116. doi: 10.1016/j.biomaterials.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).O’Connell MA, Kelleher D, Liskamp RM, Hall N, O’Neill LAJ, Long A. TNF-mediated cytotoxicity of L929 cells: Role of staurosporine in enchancement of cytotoxicity and translocation of protein kinase C isozymes. Cytokines. 1997;9:83–92. doi: 10.1006/cyto.1996.0140. [DOI] [PubMed] [Google Scholar]
- (21).Tsukamoto H, Fukudome K, Kohara J, Nakatake H, Kimoto M. Preparation of recombinant murine tumor necrosis factor-α in Escherichia coli: A rapid method to remove tags from fusion proteins by thrombin-cleavage and ion-exchange chromatography. Protein Expression Purif. 2007;56:138–144. doi: 10.1016/j.pep.2007.07.004. [DOI] [PubMed] [Google Scholar]
- (22).Nakamura K, Murray RJ, Joseph JI, Peppas NA, Morishita M, Lowman AM. Oral insulin delivery using P(MAA-g-EG) hydrogels: Effects of network morphology on insulin delivery characteristics. J. Controlled Release. 2004;95:589–599. doi: 10.1016/j.jconrel.2003.12.022. [DOI] [PubMed] [Google Scholar]
- (23).Caldorera-Moore M, Peppas NA. Micro- and nano-technologies for intelligent and responsive biomaterial-based medical systems. Adv. Drug Delivery Rev. 2009;61:1391–1401. doi: 10.1016/j.addr.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Foss AC, Goto T, Morishita M, Peppas NA. Development of acrylic-based copolymers for oral insulin delivery. Eur. J. Pharm. Biopharm. 2004;57:163–169. doi: 10.1016/S0939-6411(03)00145-0. [DOI] [PubMed] [Google Scholar]
- (25).Blanchette J, Peppas NA. Cellular evaluation of oral chemotherapy carriers. J. Biomed. Mater. Res., Part A. 2005;72A:381–388. doi: 10.1002/jbm.a.30243. [DOI] [PubMed] [Google Scholar]
- (26).Peppas NA, Wood KM, Blanchette JO. Hydrogels for oral delivery of therapeutic proteins. Expert Opin. Biol. Ther. 2004;4:881–887. doi: 10.1517/14712598.4.6.881. [DOI] [PubMed] [Google Scholar]
- (27).Lowman AM, Peppas NA. Molecular analysis of interpolymer complexation in graft copolymer networks. Polymer. 2000;41:73–80. [Google Scholar]
- (28).Torres-Lugo M, Peppas NA. Molecular design and in vitro studies of novel pH-sensitive hydrogels for the oral delivery of calcitonin. Macromolecules. 1999;32:6646–6651. [Google Scholar]
- (29).Tuesca A, Nakamura K, Morishita M, Joseph J, Peppas NA, Lowman A. Complexation hydrogels for oral insulin delivery: Effects of polymer dosing on in vivo efficacy. J. Pharm. Sci. 2008;97:2607–2618. doi: 10.1002/jps.21184. [DOI] [PubMed] [Google Scholar]
- (30).Carr DA, Gomez-Burgaz M, Boudes MC, Peppas NA. Complexation hydrogels for the oral delivery of growth hormone and salmon calcitonin. Ind. Eng. Chem. Res. 2010;49:11991–11995. doi: 10.1021/ie1008025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Morishita M, Lowman AM, Takayama K, Nagai T, Peppas NA. Elucidation of the mechanism of incorporation of insulin in controlled release systems based on complexation polymer. J. Controlled Release. 2002;81:25–32. doi: 10.1016/s0168-3659(02)00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cathou RE, Kulczycki A, Haber E. Structural features of y-immunoglobulin, antibody, and their fragments: Circular dichroism studies. Biochemistry. 1968;17:3958–3964. doi: 10.1021/bi00851a024. [DOI] [PubMed] [Google Scholar]