

Abstract
Nucleic acid-based vaccines (NAVs) are a promising alternative to conventional influenza vaccines with the potential to increase influenza vaccine availability due to their simplicity in design and rapid speed of production. NAVs can also target multiple influenza antigens and control flu variants. Traditionally NAVs have been DNA plasmids however, we are continuing to explore new methods that may enhance vaccine efficacy. Recently new focus has been on RNA cassettes as NAVs. RNA vaccines combine conceptual advantages in that they focus on delivery of only the coding cassette. However, RNA vaccines have a short half-life and cause interferon-induced fevers. Here we describe a new NAV approach where we study delivery of a linear DNA cassette [Doggybone™ linear closed DNA [(dbDNA™)] produced by an enzymatic process that yields an antigen expression cassette comprising a promoter, DNA antigen, poly A tail, and telomeric ends. This focused approach has many of the advantages of plasmid DNA as well as a minimal cassette size similar to RNA strategies. For this study, we characterized the specific CD4+ and CD8+ T cell responses and determined the hemagglutination inhibition (HI) titers induced by dbDNA™ and compared the responses with those of an optimized plasmid DNA (pDNA) vaccine encoding the same H1N1 influenza A/PR/8/34 HA gene. Immunizations with the constructs resulted in similar humoral and cellular immune responses. Both constructs induced high-titer HI antibodies and fully protected animals from lethal viral challenge. The data obtained from this study provides important validation for further development of novel vector approaches.
Keywords: doggybone, influenza, linear DNA, neutralization, plasmid DNA
Introduction
Newly reformulated seasonal vaccines need to be developed every year to protect against the point mutations that occur in the hemagglutinin (HA) and neuraminidase (NA) surface proteins of the ever-evolving influenza virus. Using the current manufacturing process, production of large quantities of the seasonal vaccine can take at least 6 months.1 However, if a reassorted virus emerges, like the 2009 H1N1 pandemic virus, development of an initial lot of vaccine can take 5 to 6 months once the strain is identified and isolated but mass production of the vaccine would require additional months.2 Therefore, there is a need for new, more efficient technologies for influenza vaccine production that will decrease manufacturing time and generate a sufficient vaccine supply to immunize at risk populations.
The production of nucleic acid-based vaccines (NAVs) represents a promising, efficient alternative to conventional influenza vaccine manufacture. The formulation of a traditional NAV requires the identification and subsequent cloning of a gene sequence of an influenza viral protein into a DNA plasmid vector.3 The plasmids are then manufactured and administered as a vaccine. Therefore, DNA vaccine manufacture eliminates the stages of production requiring vaccine virus growth and replication as with conventional vaccines and has the potential to decrease influenza vaccine manufacturing time from months to weeks.3 In addition to being non-live and non-replicating, DNA vaccines are safe and immunogenic.4 Vaccination with DNA has induced strong cellular and protective immunity against consensus H5 influenza antigens5 and H7N9 hemagglutinin antigens in mice.6 Moreover, a synthetic DNA consensus plasmid expressing hemagglutinin H5, neurominidase N1, and nucleoprotein influenza antigens induced protective humoral and cellular immunity in mice, ferrets, and non-human primates.7 Therefore, as a result of the ease of manufacture and the degree of immunogenicity of DNA, this vaccine platform is quickly emerging as an interesting approach for influenza vaccination.
One attribute of DNA that makes it particularly attractive for use as an influenza vaccine candidate is its flexibility in vaccine design. DNA vaccines are historically constructed as circularized plasmids, however, new DNA vaccine technologies are emerging in which the DNA is manufactured in a cell-free process that avoids bacterial fermentation and yields a vaccine that is structurally linear.8,9 These structurally linear, coding region only cassettes also have similarities to small RNA cassette technologies without the associated interferon stimulation driven by RNA.10,11
Recently, a novel linear DNA vector encoding an H1N1 hemagglutinin gene was enzymatically developed by Touchlight Genetics Ltd.8 This covalently closed linear DNA construct was identified as ‘Doggybone™ closed linear DNA’ (dbDNA™) based on its structural design. The linear DNA was the first rapidly-produced, plasmid-free construct synthesized through an enzymatic process that yielded a vector cassette containing only the encoded antigenic sequence, promoter, poly A tail, and telomeric ends. In an initial study, the in vivo expression and immunogenicity of the linear dbDNA™ was characterized and ELISA and induction of IFN-γ responses were reported.8
Here we build on these early studies to further characterize the specific CD4+ and CD8+ T cell responses and hemagglutination inhibition (HI) antibody titers induced by the dbDNA™ and compare the responses with those of our optimized DNA plasmid expressing the same hemagglutinin gene of H1N1 influenza A/Puerto Rico/8/1934. We report that the DNA vaccine constructs induced equivalent humoral and similar CD4+ and CD8+ T cell responses. In addition, we report that both constructs induced high-titer neutralizing antibodies that fully protected animals from lethal viral challenge. The data obtained from this study provides validation for further development of this novel DNA vector. Furthermore, since the method of synthesizing this DNA vector results in stable vectors that can be rapidly produced, use of this new manufacturing technology warrants additional study in the application of influenza vaccines.
Results
Development of the linear dbDNA™ vaccine construct
The linear dbDNA™ construct was produced using the enzymatic process depicted in Figure 1A.12 This process consisted of 2 steps; first plasmid DNA that has the sequence for the antigen flanked by telRL sites is amplified by rolling circle replication using phi29 DNA polymerase from Bacillus subtilis phage phi29, resulting in the production of long concatamers. The protelomerase TelN (from Escherichia coli phage N15) then cleaves the concatamers into strands containing a single cassette and seals the ends with a short hairpin loop.13 The construct is composed of a linear double-stranded region with an antigen expression cassette, encoding the sequences for the cytomegalovirus immediate early promoter plus enhancer, the PR8 HA gene (lacking the IgE leader sequence), and the SV40 late poly A tail, flanked by single-stranded telomere ends (Fig. 1B). In the initial round of amplification, plasmid DNA is used as a template, but this is then selectively digested with restriction enzymes and then exonuclease III. In subsequent rounds of amplification the Doggybone™ itself can be used as the template.
Figure 1.

Construction and representative expression of dbDNA™ PR8 and pDNA PR8 constructs. (A) Process of enzymatic production of dbDNA™. Rolling circle amplification of the double-stranded DNA template results in concatamers that are cleaved and joined by the protelomerase TelN to yield the covalently closed, double-stranded cassette. (B) Schematic of the linear double-stranded dbDNA™ PR8 construct with end terminal single-stranded DNA hairpins. The end product was treated with restriction enzymes and exonuclease to remove plasmid backbone sequences. (C) Schematic of pDNA PR8 construct, PR8.HA.ECRO. ECRO refers to the gene sequence containing an IgE leader sequence [e] and the target sequence is codon [c] and RNA [r] optimized [o]. The sequence of PR8 was cloned into the pVax1 mammalian expression vector. The CMV promoter, HA gene, BGH poly A signal, kanamycin resistance gene, and pUC origin are shown. (D) Representative in vitro expression of the dbDNA™ PR8 and pDNA PR8 constructs. Expression was confirmed using transfected RD cells and a HA-tagged antibody. An empty vector (pVax) was used as a negative control. Results were analyzed with confocal imaging. Expression is indicated by fluorescein isothiocyanate (FITC) staining (green).
Expression of linear dbDNA™ PR8 and pDNA PR8 vaccines
To determine expression of the DNA constructs, an indirect immunofluorescence assay was performed. Since each DNA construct would be delivered intramuscularly into the tibialis anterior muscles of the mice, we wanted to show that the DNA plasmids were capable of transfecting a mammalian muscle cell line. To accomplish this DNA transfection, we chose Rhabdomyosarcoma (RD) muscle cells. Each DNA construct was individually transfected into RD muscle cells. As a negative control, transfection was also performed with an empty vector backbone, pVax. Post-transfection, immunofluorescent staining was carried out using a hemagglutinin-tagged antibody. Plasmid expression was confirmed with a FITC-labeled secondary antibody (green staining) (Fig. 1D). Both DNA constructs were expressed in vitro. Expression was not detected in pVax- transfected cells. In vivo expression of dbDNA™ was previously reported.8
Vaccination with linear and plasmid DNA induces comparable humoral immune responses
After confirming the cellular expression of both DNA constructs, we wanted to evaluate whether the antibody responses induced by the dbDNA™ were comparable to those induced by pDNA. Mice were immunized 3 times, 3 weeks apart with dbDNA™ PR8 or pDNA PR8 and serum analysis was performed 3 weeks after each immunization to determine the antibody responses induced by each vaccination (Fig. 2). Since the influenza HA gene must undergo cleavage to form HA1 and HA2 to yield infectious virus, we chose to evaluate antibody responses to an infectious viral protein, H1N1 influenza A/PR/8/34 HA1, by ELISA. As shown in Figure 3, we detected high-titer antibody responses to both DNA vectors. In addition, the antibody responses induced by the DNA were dependent on the number of immunizations. The group geometric mean anti-A/Puerto Rico/8/34 HA1 specific endpoint titers induced by dbDNA PR8™ and pDNA PR8 were (7.2 × 102 and 1.9 × 103), (1.8 × 105 and 3.2 × 105), and (3.2 × 105 and 3.5 × 105) after one, 2, and 3 immunizations, respectively. These results demonstrate the ability of dbDNA™ PR8 to induce high-titer antibody responses that are comparable to those induced by pDNA PR8.
Figure 2.

Study timeline. BALB/c mice immunized with either dbDNA™ PR8 or pDNA PR8 received the treatments listed at the indicated weeks.
Figure 3.

DNA vaccination induces robust antibody responses. Comparison of the antibody responses induced by each vaccination. Absorbances and geometric mean endpoint titers against A/PuertoRico/8/34 are shown for both the dbDNA™ PR8 (A and C) and pDNA PR8 (B and D) constructs. Each vaccination is noted as Dose 1, Dose 2, or Dose 3.
DNA vaccination induces strong hemagglutination inhibition antibody titers
After determining that both DNA constructs induced high-titer antigen-specific antibodies, we wanted to evaluate whether the constructs generated HI antibodies. We used a standard Hemagglutination Inhibition (HI) assay to test the ability of immune sera to inhibit red blood cell agglutination by the H1N1 influenza A/PR/8/34 strain. Serum samples collected from the mouse challenge groups (n = 10 / group) 3 weeks after the final vaccination and prior to viral challenge and were used in the assay. The average reciprocal HI antibody titer induced by vaccination with dbDNA™ PR8 and pDNA PR8 were 1:2688 and 1:2560, respectively (Fig. 4A). In addition, sera from naïve mice did not neutralize virus. These data show that both DNA constructs induced protective HI antibody titers (≥1:40).
Figure 4.
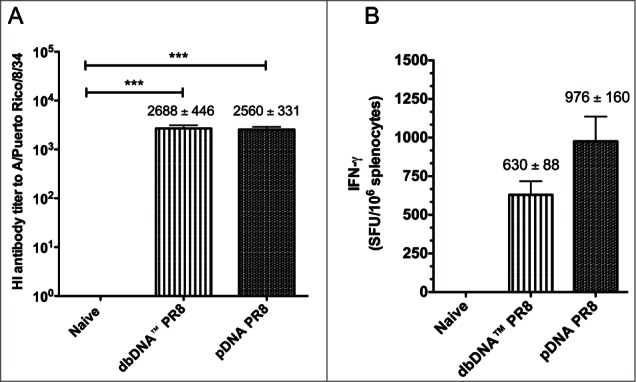
Comparison of host responses to DNA vaccination. (A) Hemagglutination inhibition (HI) antibody titers from the challenge groups of mice (n = 10/group) 3 weeks after the final immunization and prior to viral challenge. (B) The number of IFN-γ secreting splenocytes 3 weeks after the final vaccination. Splenocytes were stimulated with H1HA pooled peptides and IFN-γ secretion was reported as spot forming units (SFU) per million splenocytes. Group mean ± SEM are reported. ***, P < 0.0005 for DNA HI antibody titers compared to naive.
DNA vaccination induces multifunctional antigen-specific T-cell responses
Since a cell-mediated immune response is needed to target live influenza virus infection,14 we investigated the ability of dbDNA™ PR8 and pDNA PR8 to induce antigen-specific cellular immune responses (Fig. 4B and Fig. 5). Mice from the cellular immunogenicity study (n = 5 / group) were immunized 3 times, 3 weeks apart with either dbDNA™ PR8 or pDNA PR8. Splenocytes were harvested 3 weeks after the final vaccination and cells were used in an IFN-γ ELISpot assay. As shown in Figure 4B, the average response to antigenic stimulation against 4 H1HA pooled peptides in mice immunized with either dbDNA™ PR8 or pDNA PR8 was 630 ± 88 and 976 ± 160 SFU/106 splenocytes, respectively. Naïve wells contained only minimal background spots.
Figure 5.

Cytokine frequencies and phenotypic profiles of specific CD4+ and CD8+ T cells following DNA immunizations. Cytokine recall responses to dbDNA™ PR8 and pDNA PR8 were measured 3 weeks after the final immunization by ICS and flow cytometry. Multiparameter flow cytometry was used to determine the percentages of multifunctional CD4+ T cell (A–C) and CD8+ T cell (D–F) cytokines. The graphs show the percentages of each cytokine subpopulation to H1HA pooled consensus peptide stimulation. Background staining from cells stimulated with medium alone has been subtracted. Data represent the mean ± SEM of 5 mice per group. Cytokine responses for animals vaccinated with dbDNA™ PR8 or pDNA PR8 were compared to naïve. ***, P < 0.0005; **, P < 0.005; *, P < 0.05.
After determining that both DNA constructs induced strong IFN-γ responses, we characterized the phenotype and cytokine profiles of antigen-specific T cells (Fig. 5). Because multifunctional CD4+ and CD8+ antigen-specific T cells play an important role in providing protective immunity from lethal influenza virus infection,14 we measured the percentages of polyfunctional CD4+ T cell (Fig. 5A–C) and CD8+ T cell (Fig. 5D–F) cytokine responses induced by both DNA constructs. Additional splenocytes harvested from mice in the cellular immunogenicity study were stimulated in vitro with H1HA pooled peptides and the production of IFN-γ, TNF-α, and IL-2 by CD4+ and CD8+ T cells was quantified. Immunization with either DNA construct resulted in CD4+ and CD8+ antigen-specific T-cells that produced all 3 cytokines. The percentages of CD4+ T cells producing total IFN-γ (dbDNA™ PR8 0.10%; pDNA PR8: 0.13%), total TNF-α (dbDNA™ PR8: 0.14%; pDNA PR8: 0.17%), and total IL-2 (dbDNA™ PR8: 0.08%; pDNA PR8: 0.10%) (Fig. 5A–C). The percentages of CD8+ T cells producing total IFN-γ (dbDNA™ PR8: 0.51%; pDNA PR8: 0.70%), total TNF-α (dbDNA™ PR8: 0.43%; pDNA PR8: 0.64%), and total IL-2 (dbDNA™ PR8: 0.10%; pDNA PR8: 0.14%) (Fig. 5D–F). Although the percentages of CD4+ and CD8+ T cells that produced all 3 cytokines were similar when responses of animals immunized with dbDNA™ PR8 were compared to those immunized with pDNA PR8, slightly higher CD4+ and CD8+ T cell responses were observed in animals that received the plasmid form of the vaccine. In addition, we observed higher percentages of CD8+ T cells secreting antiviral cytokines than CD4+ T cells in animals that received either DNA construct. All T-cell responses were significantly higher than those of naïve mice, indicating that DNA vaccination induces potent, multifunctional cellular immune responses against influenza infection.
DNA vaccination provides protective immunity from lethal influenza challenge
Given that vaccination with both DNA constructs elicited strong HI antibody titers, we wanted to evaluate whether the vaccines would provide protective immunity from lethal influenza infection in vivo. Since there are some PR8 strains that differ by a few amino acids in critical binding sites, we chose a challenge virus (PR8 Cambridge) that is antigenically identical to our optimized PR8 encoded HA gene.15 Mice from the challenge groups (n = 10 / group) were intranasally challenged with 50 LD50 PR8 Cambridge and monitored for weight loss and survival for 16 days post-challenge (Fig. 6). A group of unchallenged mice (n = 10) received PBS to show that virus diluent was non-toxic to animals (data not shown). All DNA immunized animals maintained their body weight and were fully protected against lethal challenge. Naïve animals lost 20% of their body weight by day 6, indicating lethal infection, and were humanely sacrificed. Unchallenged mice that were administered PBS survived the study time course indicating that the PBS virus diluent was non-toxic. These data provide evidence that vaccination with either linear or plasmid DNA is effective at inducing protective immunity against influenza virus infection.
Figure 6.

Protection from H1N1 A/Puerto Rico/8/34 virus challenge in DNA immunized mice. Percentage body weight loss following lethal viral challenge with 50 LD50 PR8 Cambridge virus and Kaplan-Meier survival curve showing percentage survival of mice from each group. (A and B) Percentage body weight loss and survival for dbDNA™ PR8 immunized mice. (C and D) Percentage body weight loss and survival for pDNA PR8 immunized mice. Group mean and SEM are reported. ###, P < 0.0005 for dbDNA™ PR8 body weight (%) compared to naïve. ***, P < 0.0005 for pDNA PR8 body weight (%) compared to naïve.
Discussion
The rapid production and immunogenicity of DNA make this vaccine platform a promising alternative to the manufacture and use of many conventional vaccines. Additional modification of the DNA cassettes may further enhance their immunogenicity.5-7,16 An emerging new method in DNA vaccine design which allows cellular targeting of DNA and additional strategies to improve the immunogenicity of the platform is the production of linear DNA.16 Linear DNA cassettes mimic aspects of mRNA vaccines in that they are highly focused on delivery of coding cassettes but this approach has advantages over the RNA platform in the amplification of the mRNA signal produced as well as in avoiding the induction of unwanted interferon responses.10,11 The use of a linear DNA encoding influenza antigens was reported to induce protective immune responses comparable to immunization with plasmid DNA.9 Here we examined the use of linear DNA cassettes created by a novel enzymatic strategy for generation of a covalently closed linear DNA vector (Doggybone™ DNA) developed by Touchlight Genetics Ltd. In an initial study, the Doggybone™ vector was reported to induce humoral and cellular immune responses equivalent to plasmid DNA.8 We further evaluated these new linear candidate vaccines and characterized specific CD4+ and CD8+ T cell and neutralizing antibody responses. We compared the responses to those of our optimized plasmid DNA vector encoding an identical H1N1 influenza A/PR/8/34 HA gene insert.
We evaluated the in vitro expression of the Doggybone™ vector and our plasmid vector. We used an indirect immunofluorescence assay and probed for cellular transfection using a HA-tagged antibody. The immunofluorescence assay is a sensitive assay that allowed us to detect the presence and localization of the hemagglutinin protein within the muscle cells. We found that both DNA vectors were expressed in the cells. These results show that the structural design of DNA constructs does not limit cellular expression. Due to an intensive, comparative in vitro and in vivo assessment of expression of the Doggybone™ construct and a traditional plasmid DNA construct in the initial Doggybone™ study,8 no further analysis of Doggybone™ expression was required for this study. One concern of the initial study was the size of the Doggybone™ DNA vector. Since the Doggybone™ vector is smaller than pDNA, equal masses of the 2 constructs will result in a higher number of gene copies with Doggybone™ DNA compared to pDNA. Therefore, Walters et al. performed an in vivo luciferase expression analysis of the Doggybone™ vector and the plasmid vector in a mass-matched and molarity-matched study. In the study of molarity, they found that there was a trend toward reduced expression of the Doggybone™ vector. However, Walters et al. concluded that the levels of gene expression of the Doggybone™ vector and plasmid vector were comparable. Based on the expression results from the initial study, we were confident that a mass-matched study of dbDNA™ and pDNA would yield similar expression levels and our in vitro results were consistent with this hypothesis.
After confirming expression of both DNA constructs, we compared the antibody-mediated immune responses induced by vaccination. The clear correlate of protective immunity against influenza infection is reactive antibodies, particularly hemagglutination inhibition antibodies. Since the production of antibodies against the post-transcriptionally cleaved HA surface protein targeting the HA1 region can effectively enhance viral neutralization, we first compared the antibody response against H1N1 influenza A/PR/8/34 HA1 induced by vaccination with the 2 structurally different DNA constructs. Vaccination with the DNA constructs yielded similar high-titer antibody responses that were dependent on the number of administrations. These results indicate that immunization with dbDNA™ induces potent antibody responses that are comparable to those induced by plasmid DNA.
The protective correlate of influenza viral immunity is the level of hemagglutination inhibition antibodies. A titer of at least 1:40 is considered the benchmark for a protective influenza vaccine. This is the minimum titer that is associated with a 50% reduced risk of infection.17,18 To that end, we compared the levels of neutralizing antibodies induced by the DNA vaccinations with a HI assay. Vaccination with the DNA constructs induced similar HI antibody titers to H1N1 influenza A/PR/8/34. In addition, the virus HI antibody titers were about 66X higher than the HI correlate for protective immunity, HI ≥ 1:40. These results provide evidence that both antigenic forms of DNA were capable of eliciting a protective immune response to influenza H1N1 infection. Our data along with the HI data of other DNA vaccines6,7 further support the effectiveness of the DNA vaccine platform to provide protective immunity from influenza. However, to our knowledge this is the first report of DNA vaccines that are capable of inducing HI titers greater than 1:2000. This observation of high HI titers supports the need for additional study of the optimized dbDNA™ and plasmid DNA as potential influenza vaccine candidates to provide protective immunity against lethal influenza A viruses.
Although viral neutralizing antibodies primarily mediate immune protection against influenza, the role of multifunctional CD4+ and CD8+ antigen-specific T cells in providing protective immunity from influenza infection is currently being evaluated.14 The ability of CD4+ and CD8+ T cells to secrete antiviral cytokines such as IFN-γ and TNF-α may enhance antiviral immunity.14 Since an ideal influenza vaccine candidate should elicit a strong, polyfunctional cell-mediated immune response, we wanted to determine and compare the cytokine frequencies and phenotypic profiles elicited by our DNA vaccines. We quantified the production of IFN-γ, TNF-α, and IL-2 by CD4+ and CD8+ T cells from mice immunized with the DNA constructs. We found that all 3 cytokines were secreted by CD4+ and CD8+ T cells of DNA immunized animals, although the higher cytokine percentages were associated with animals receiving plasmid DNA. In addition, there were higher percentages of CD8+ T cells secreting antiviral cytokines than CD4+ T cells in all DNA immunized animals. This data illustrate that the linear cassette and plasmid both exhibit unique immune profiles. Antigen-specific CD8+ T cell responses to influenza are reported to enhance clearance of the virus and promote recovery from secondary infections in mice that lack humoral immunity. 19-22 The high percentages of CD8+ T cells induced by our DNA vaccines may enhance antiviral immunity against influenza A. In addition, since CD4+ cells provide T cell help, the antigen-specific T cells induced by both DNA formulations may contribute to the activation of B cells to produce antigen-specific antibodies. These results indicate that both DNA vaccine constructs are capable of inducing potent, cellular immune responses against influenza infection.
Our study demonstrates that immunization with closed linear DNA induces protective immunity to influenza infection and the response induced is comparable to those of plasmid DNA. The immunogenicity of the dbDNA™ further supports the future study of the enzymatically-produced construct as an alternative NAV formulation that may have particular advantages over RNA-based approaches. The rapid manufacturing process of dbDNA™merits additional study as well.
Materials and Methods
Preparation and design of dbDNA™PR8
The proTLx™ expression plasmid consisted of the CMV Immediate early promoter plus enhancer, a multiple cloning site and an SV40 late polyadenylation signal all flanked by 2 telRL sequences, the site of protelomerase TelN recognition and cleavage. The plasmid backbone contained an ampicillin resistance gene and the pUC origin of replication. The hemagglutinin gene (without the IgE leader sequence) from H1N1 influenza A/PR/8/34 was PCR-amplified from pDNA PR8 from cloned (adding a GCCACC Kozak sequence and HindIII and EcoRI sites) into the HindIII and EcoRI sites on the proTLx™ base plasmid. The enzymes used in the plasmid construction were sourced from New England Biolabs. Plasmids were extracted using Miniprep kit (Qiagen). The template plasmid was maintained in recombinase-deficient E. coli strain. The template plasmid was verified by Restriction Endonuclease digestion and Sanger sequencing.
Purification of TelN protelomerase
The gene encoding protelomerase TelN, from E. coli phage N15, was cloned into pQE-80L kan (Qiagen) under the control of the IPTG-inducible promoter from phage T5. The enzyme was overproduced in E. coli BL21 DE3 cells (Life Technologies), with an N-terminal 6 × Histidine tag. Cell disruption and clarification was achieved using a cell disrupter (2 passes × 18 k psi using a TS Series, Constant Systems) followed by centrifugation (30 min at 14000 g at 4°C). The supernatant was filtered through a 0.2 μm filter and loaded onto a 16/10 Hisprep FF column (GE Healthcare) pre-equilibrated with 20 mM Tris pH 7.4 150 mM NaCl. Fractions containing the TelN protein were loaded onto a 16/10 Hiprep Heparin FF column (GE Healthcare), the protein was eluted using a linear gradient of NaCl. The eluted protein was buffer exchanged using a Hiprep 26/10 desalting column (into 10 mM Tris HCl pH 7.4, 75 mM NaCl, 50% (v/v) glycerol, 1mM DTT and 0.1 mM EDTA) and stored at a concentration of approximately 15 µM at −20°C.
Preparation of closed linear DNA
Plasmid was prepared using a maxiprep kit (Sigma) and used as a template for rolling circle amplification by the method described in the patent EP2391731. The template plasmid containing the PR8 cassette flanked by telRL sites was mixed with custom primers (50 μM), and the template was denatured by heating to 95 °C. To initiate rolling circle amplification from the denatured template, the reaction was first mixed with reaction buffer (30 mM Tris-HCl pH 7.4, 5 mM (NH4)2SO4, 30 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol) and then 2 mM dNTPs (Bioline) were added together with 4000 units of Phi29 DNA polymerase (Lucigen) and 4 units of thermostable pyrophosphatase (Enzymatics). Upon mixing, the reaction was incubated at 30°C for 18 h with custom primers (50 μM) and 2 mM dNTPs. Concatameric DNA was processed by the addition of 2 μM TelN to produce dbDNA™ and a backbone doggybone. The digest mixture was cleaned from reaction components by the addition of 2M NaCl and precipitated using 5% PEG 8000 (Promega). DNA was pelleted by centrifuging at 14,000g for 10 mins. DNA pellets were resuspended in 20 ml NEB buffer 2 and plasmid backbone was removed by incubating the TelN digest mixture with 200U/ml ApaLI (NEB) and 400U/ml ExoIII (Enzymatics) overnight at 37°C. dbDNA™ was partially cleaned from the reaction components by performing a 3% PEG 8000 precipitation (removal of proteins) and 6 % PEG precipitation to pellet dbDNA™ from dNMPs and oligonucleotides. This was repeated to increase efficiency and the final 6% pellet resuspended in sterile H2O. The sample was then diluted to a final volume of 20 ml in 20 mM Tris HCl pH 7.4 and loaded onto a column packed with POROS 50 HQ anion exchange (Life Technologies) on an Akta Purifier 10 system. The dbDNA™ was eluted using a linear 0–2M NaCl gradient. Fractions containing the dbDNA™ were then buffer exchanged in sterile H2O using size exclusion (Hiprep 26/60 Sephacryl S-200 HR, GE Healthcare). The resulting elutions were concentrated using 30 k MWCO Amicon columns (Millipore) to a final concentration of 3.6mg/ml.
Preparation and design of pDNA PR8
To design a plasmid DNA vaccine targeting the hemagglutinin (HA) of H1N1 influenza A/PR/8/34 (PR8), the HA gene sequence of PR8 was identified and codon and RNA optimized for expression in humans using GeneOptimizer® sequence analysis software (Life Technologies). The optimized H1N1 influenza A/PR/8/34 HA gene was cloned into the pVax expression vector, controlled by the cytomegalovirus immediate-early promoter, between the EcoR1 and Not1 sites. The construct was amplified by Aldevron (Fargo, ND) and the resulting purified DNA plasmid was formulated with sterile water and used in animal vaccinations.
Indirect immunofluorescent assay
An indirect immunofluorescent assay was used to confirm expression of the dbDNA™ PR8 and pDNA PR8 vectors. Rhabdomyosarcoma (RD) cells (2 × 105 cells) (ATCC, CCL−136) were seeded in 2-chamber tissue culture treated glass slides (BD Falcon) and transfected with 6 μg/well of either dbDNA™ PR8 or pDNA using Turbofectin (Origene, TF81001). As a negative experimental control, RD cells were transfected with pVAX (6 μg/well). Transfected cells were maintained in culture in Dulbecco's Modified Eagle's Medium (DMEM; Gibco-Invitrogen, 11965-084) supplemented with 10% fetal calf serum (FBS) (Atlas Biologicals, Inc.., F0500-A) and 1% penicillin-streptomycin solution (10,000 U/ml) (Invitrogen, Inc.., 15140-122). Forty-eight hours post-transfection, cells were fixed with 2% paraformaldehyde, washed in 1X phosphate buffered saline (PBS) (Gibco-Invitrogen, 14190-136) and incubated with goat anti-mouse HA tag antibody (Abcam, ab18181) 1:100 dilution in primary standard solution (PSS) (0.1% BSA, 0.2% saponin, and 0.02% sodium azide in PBS) (37°C, 1.5 h). Cells were washed in PBS and incubated with goat polyclonal secondary antibody to mouse IgG conjugated to fluorescein isothiocyanate (FITC) (Abcam, ab6785) 1:100 dilution in PSS for 1 h at room temperature. After washing, cell nuclei were counterstained with Hoechst reagent (Sigma-Aldrich, H6024) and slides were mounted with fluoromount G (Electron Microscopy Sciences, 17984-25). Expression of the DNA constructs was confirmed by confocal imaging. Confocal images were acquired using the Zeiss LSM 510 NLO/META Confocal Microscope at the Cell and Developmental Biology Microscopy Core, University of Pennsylvania, PA, USA.
Immunization of mice
Female BALB/c mice (6 to 8 weeks of age) were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were housed in a temperature-controlled, light-cycled facility and received food and water ad libitum. All experiments were performed in accordance with the guidelines of the National Institutes of Health (NIH) (Bethesda, MD) and the University of Pennsylvania (Philadelphia, PA) Institutional Animal Care and Use Committee (IACUC #804321).
In a previous dosage study (data not shown), the immunogenicity of dbDNA™ PR8 and pDNA PR8 administered at 5 µg, 10 µg, 25 µg, and 50 µg was evaluated. From that initial study, 25 µg of dbDNA™ PR8 and pDNA PR8 was determined as the optimal dosage and was used for further study. For the humoral immunogenicity/challenge study, 40 mice were divided into 4 groups (n = 10/group) and received no vaccine (Naïve), no vaccine (no viral challenge), 25 µg dbDNA™ PR8, or 25 µg pDNA PR8. All vaccinated animals received 3 immunizations spaced 3 weeks apart. Animals received intramuscular injections into the tibialis anterior muscle. Intramuscular injection was immediately followed by electroporation (EP). Briefly, mice received 2 constant-current pulses of 0.2 A delivered through a triangular 3-electrode array consisting of 26-gauge solid stainless steel electrodes. Pulses were 52 ms in length separated by a one second delay. All in vivo electroporation procedures were performed using the CELLECTRA® 3P electroporation device (Inovio Pharmaceuticals, Inc., Plymouth Meeting, PA). Sera samples were collected prior to each subsequent immunization to monitor the dose response.
For the cellular immunogenicity study, 15 mice were divided into 3 groups (n = 5/group) and received no vaccine (Naïve), 25 µg dbDNA™ PR8, or 25 µg pDNA PR8. All vaccinated animals received 3 immunizations spaced 3 weeks apart. Animals received intramuscular injections into the tibialis anterior muscle followed by EP using the CELLECTRA® 3P device. All animals were humanely sacrificed 3 weeks after the final immunization and splenocytes were harvested to determine the cellular immune response.
H1N1 influenza A/PR/8/34 HA-specific serum antibody measurements
To measure H1N1 influenza A/PR/8/34 HA-specific serum IgG antibodies in immunized and control animals, we performed an ELISA. EIA/RIA 96 well flat-bottom plates (Corning Life Sciences, 3590) were coated with 100 µl/well 1X PBS containing 0.3 μg/ml recombinant HA1 (H1N1/PR8/34) (Immune Technology Corp., IT-003-0010p). Plates were washed with PBS-T (0.05% Tween 20 in PBS) and blocked with 0.5% BSA in PBS-T at room temperature for 2 h. Plates were incubated with serial dilutions of immunized and naïve sera in 0.2% BSA in PBST (37°C, 1.5 h). Plates were washed with PBS-T and incubated with goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, Inc., sc-2055) at a 1:5000 dilution in 0.2% BSA in PBS-T. After washing, the enzyme substrate SigmaFAST o-phenylenediamine dihydrochloride (OPD) (Sigma-Aldrich, P1987) was added. The development was stopped with the addition of 100 μl 1N H2SO4 and optical density was determined at 450 nm. Endpoint titers were determined as previously described.23 Endpoint titers are reported as the reciprocal of the last dilution that had an absorbance above the upper prediction limit.
Hemagglutination inhibition assay
To determine the hemagglutination inhibition titer to H1N1 influenza A/PR/8/34, sera was harvested from mice in the humoral immunogenicity/challenge study 3 weeks after the final immunization. Sera samples were treated with receptor-destroying enzyme (RDE) at a 1:3 ratio at 37°C for 18 h followed by complement inactivation at 56°C for 45 min. RDE-treated sera were diluted 1:10 in PBS in a 96-well V-bottom microtiter plate and further diluted 2-fold down the plate. Four hemagglutinating doses of PR8 Cambridge virus were added to each well and the serum-virus mixture was incubated at room temperature for 1 h. Following incubation, 50 μl of turkey red blood cells (0.5% cells in 0.85% NaCl) were added to each well and incubated at room temperature for 45 min. The HI antibody titer was scored as the reciprocal of the highest dilution that prevented red blood cell agglutination.
Splenocyte harvest
To evaluate the cellular immune response induced by vaccination with dbDNA™ PR8 and pDNA PR8 vectors, mice from the cellular immunogenicity study (as previously described) were humanely euthanized 3 weeks after the final vaccination and spleens were harvested from each animal. Spleens were placed in complete RPMI 1640 medium (Mediatech, MT10-040-CM) containing 10% fetal calf serum (FCS), 1% penicillin-streptomycin (10,000U/ml) (Invitrogen), and 1X β-mercaptoethanol (Invitrogen, 21985-023). Spleens were disrupted using a Stomacher machine (Steward Laboratory Systems, Bohemia, NY) and the cellular product was strained using a 40 μm cell strainer (BD Biosciences). Red blood cells were lysed with ACK lysis buffer (Lonza, 10-548E). The remaining cells were washed with 1X PBS, resuspended in complete RPMI medium, restrained, and used in IFN-γ ELISpot and intracellular cytokine staining (ICS) assays.
IFN-γ ELISpot assay
To detect and quantify the amount of IFN-γ secreting mouse splenocytes, harvested cells were used in an IFN-γ ELISpot assay. The assay was performed using the Mouse IFN-γ Development Module (R&D Systems, SEL485) according to the manufacturer's instructions. Mouse splenocytes were resuspended in complete RPMI medium and plated at a concentration of 2 × 105 cells/well. A set of peptides, each containing 15 aa residues overlapping by 11 aa spanning the entire protein consensus sequences of H1HA (A/Taiwan/1/86, A/Bayern/07/95, A/Texas/36/91, A/Beijing/262/95, A/New Caledonia/20/99, A/Solomon Islands/03/06, A/Brisbane/59/2007, A/Puerto Rico/8/34, A/South Carolina/1/18, A/California/07/09, and A/Mexico/InDRE4487/09) were synthesized from GeneScript (Piscataway, NJ). The set of peptides was pooled to make a concentration of 2 μg/ml per peptide and divided into 4 pools for use as stimulating antigens. As a positive control, cells were stimulated with 5 mg/ml Concanavalin A (Sigma-Aldrich, C5275). Complete RPMI medium was used as a negative control. Color development was performed using the ELISPOT Blue Color Module (R&D Systems, SEL002) according to the manufacturer's directions. An automated CTL Analyzer (Cleveland, OH) was used to count spots. The number of spot forming units (SFU) was reported as SFU/1 × 106 splenocytes.
Intracellular cytokine staining and flow cytometry
Intracellular staining was performed as previously described.24 For this study, the following antibodies were used for surface staining: LIVE/DEAD Fixable Violet Dead Cell stain kit (Invitrogen), CD19 (V450; clone 1D3; BD Biosciences), CD4 (FITC; clone RM4-5; BD Biosciences), CD8 (APC-Cy7; clone 53–6.7; Abcam). For intracellular staining the following antibodies were used: IFN-γ (APC; clone XMG1.2; Biolegend), TNF-α (PE; clone MP6-XT22; ebioscience), IL-2 (PeCy7; clone JES6-SH4; ebioscience), CD3 (PerCP/Cy5.5; clone 145-2C11; Biolegend). All data were collected using a LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR) and SPICE v5.2 (free available from http://exon.niaid.nih.gov/spice/). Boolean gating was performed using FlowJo software to examine the polyfunctionality of the T cells from vaccinated animals. For flow cytometry, cells were gated on singlets using SSC-H by SSC-A followed by gating on LIVE-DEAD (dump channel), CD3+ CD4+ CD8− T and CD3+ CD8+ CD4− T cells to examine the CD4+ and CD8+ T-cell populations secreting IFN-γ, TNF-α, and IL-2 cytokines. 1 × 106/wells cells were stimulated with H1HA consensus pooled peptides for 5 hours and 500,000 events were collected by the LSRII.
Viral challenge
Four weeks following the last immunization, challenge study mice were anaesthetized with isofluorane (Abbott Laboratories, 05260-05) and intranasally challenged with 50 LD50 of the PR8 Cambridge strain of H1N1 influenza A/PR/8/34 virus in a 50 μl volume. Post-challenge, animals were weighed daily for 16 days and monitored for symptoms of viral infection. Animals that lost 20% of their pre-challenge body weight were immediately euthanized. All surviving animals were humanely euthanized after 16 d.
Statistical analysis
One-way analysis of variance (ANOVA) followed by Dunnett's Multiple Comparison post-test was used to compare the cellular immune responses and HI antibody titers between naïve mice and mice immunized with either the dbDNA™ PR8 or pDNA PR8 construct. Two-way ANOVA followed by Bonferroni post-tests was used to compare the percentage of body weight loss of viral challenged naïve mice to immunized mice. Differences were considered statistically significant at P < 0.05.
Disclosure of Potential Conflicts of Interest
D.B.W. has grant funding, participates in industry collaborations, receives speaking honoraria, and collects fees for consulting. He serves on scientific review committees and advisory boards. Remuneration includes direct payments, stock, or stock options. In the interest of disclosure, he therefore notes potential conflicts associated with this work with Pfizer, Bristol Myers Squibb, Inovio, Touchlight, oncosec, Merck, VGXI, and possibly others. Licensing of technology from his laboratory has created over 100 jobs in the private sector in the biotech/pharma industry. The other authors declare no competing financial interests.
Acknowledgments
The authors thank the laboratory of Dr. Scott E. Hensley for generously supplying the PR8 Cambridge virus stock. We thank the University of Pennsylvania Cell and Developmental Biology (CDB) Microscopy Core staff for their generous technical assistance with microscopy imaging.
References
- 1.(CDC) CfDCaP Selecting the Viruses in the Seasonal Influenza (Flu) Vaccine. Seasonal Influenza (Flu), 2014 [Google Scholar]
- 2.(WHO) WHO Pandemic (H1N1) 2009 briefing note 7. Global Alert and Response (GAR), 2009 [Google Scholar]
- 3.Morrow MP, Weiner DB. DNA drugs come of age. Sci Am 2010; 303:48-53; PMID:20583666; http://dx.doi.org/ 10.1038/scientificamerican0710-48 [DOI] [PubMed] [Google Scholar]
- 4.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Gen 2008; 9:776-88; PMID:18781156; http://dx.doi.org/ 10.1038/nrg2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laddy DJ, Yan J, Corbitt N, Kobasa D, Kobinger GP, Weiner DB. Immunogenicity of novel consensus-based DNA vaccines against avian influenza. Vaccine 2007; 25:2984-9; PMID:17306909; http://dx.doi.org/ 10.1016/j.vaccine.2007.01.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan J, Villarreal DO, Racine T, Chu JS, Walters JN, Morrow MP, Khan AS, Sardesai NY, Kim JJ, Kobinger GP, et al.. Protective immunity to H7N9 influenza viruses elicited by synthetic DNA vaccine. Vaccine 2014; 32:2833-42; PMID:24631084; http://dx.doi.org/ 10.1016/j.vaccine.2014.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laddy DJ, Yan J, Kutzler M, Kobasa D, Kobinger GP, Khan AS, Greenhouse J, Sardesai NY, Draghia-Akli R, Weiner DB. Heterosubtypic protection against pathogenic human and avian influenza viruses via in vivo electroporation of synthetic consensus DNA antigens. PloS one 2008; 3:e2517; PMID:18575608; http://dx.doi.org/ 10.1371/journal.pone.0002517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters AA, Kinnear E, Shattock RJ, McDonald JU, Caproni LJ, Porter N, Tregoning JS. Comparative analysis of enzymatically produced novel linear DNA constructs with plasmids for use as DNA vaccines. Gene Ther 2014; 21:645-52; PMID:24830436; http://dx.doi.org/ 10.1038/gt.2014.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kendirgi F, Yun NE, Linde NS, Zacks MA, Smith JN, Smith JK, McMicken H, Chen Y, Paessler S. Novel linear DNA vaccines induce protective immune responses against lethal infection with influenza virus type A/H5N1. Hum Vaccin 2008; 4:410-9; PMID:18443425; http://dx.doi.org/ 10.4161/hv.4.6.6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollard C, Rejman J, De Haes W, Verrier B, Van Gulck E, Naessens T, De Smedt S, Bogaert P, Grooten J, Vanham G, et al.. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther 2013; 21:251-9; PMID:23011030; http://dx.doi.org/ 10.1038/mt.2012.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desmet CJ, Ishii KJ. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol 2012; 12:479-91; PMID:22728526; http://dx.doi.org/ 10.1038/nri3247 [DOI] [PubMed] [Google Scholar]
- 12.Hill V. Production of Closed Linear DNA. WO 2010086626 A1. 2010 [Google Scholar]
- 13.Heinrich J, Schultz J, Bosse M, Ziegelin G, Lanka E, Moelling K. Linear closed mini DNA generated by the prokaryotic cleaving-joining enzyme TelN is functional in mammalian cells. J Mol Med 2002; 80:648-54; PMID:12395149; http://dx.doi.org/ 10.1007/s00109-002-0362-2 [DOI] [PubMed] [Google Scholar]
- 14.Thomas PG, Keating R, Hulse-Post DJ, Doherty PC. Cell-mediated protection in influenza infection. Emerg Infect Dis 2006; 12:48-54; PMID:16494717; http://dx.doi.org/ 10.3201/eid1201.051237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982; 31:417-27; PMID:6186384; http://dx.doi.org/ 10.1016/0092-8674(82)90135-0 [DOI] [PubMed] [Google Scholar]
- 16.Zanta MA, Belguise-Valladier P, Behr JP. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci U S A 1999; 96:91-6; PMID:9874777; http://dx.doi.org/ 10.1073/pnas.96.1.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg 1972; 70:767-77; PMID:4509641; http://dx.doi.org/ 10.1017/S0022172400022610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Jong JC, Palache AM, Beyer WE, Rimmelzwaan GF, Boon AC, Osterhaus AD. Haemagglutination-inbibiting antibody to influenza virus. Dev Biol (Basel), 2003; 115:63-73; PMID:15088777 [PubMed] [Google Scholar]
- 19.Bender BS, Croghan T, Zhang L, Small PA Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 1992; 175:1143-5; PMID:1552285; http://dx.doi.org/ 10.1084/jem.175.4.1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou S, Doherty PC, Zijlstra M, Jaenisch R, Katz JM. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J Immunol 1992; 149:1319-25; PMID:1354233 [PubMed] [Google Scholar]
- 21.Graham MB, Braciale TJ. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J Exp Med 1997; 186:2063-8; PMID:9396777; http://dx.doi.org/ 10.1084/jem.186.12.2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epstein SL, Lo CY, Misplon JA, Bennink JR. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol 1998; 160:322-7; PMID:9551987 [PubMed] [Google Scholar]
- 23.Frey A, Di Canzio J, Zurakowski D. A statistically defined endpoint titer determination method for immunoassays. J Immunol Methods 1998; 221:35-41; PMID:9894896; http://dx.doi.org/ 10.1016/S0022-1759(98)00170-7 [DOI] [PubMed] [Google Scholar]
- 24.Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, Yan J, Morrow MP, Weiner DB. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res 2014; 74:1789-800; PMID:24448242; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2729 [DOI] [PMC free article] [PubMed] [Google Scholar]