

Abstract
Sclerostin inhibits bone formation mostly by antagonizing LRP5/6, thus inhibiting Wnt signaling. However, experiments with genetically modified mouse models suggest that a significant part of sclerostin-mediated inhibition of bone formation is due to interactions with other binding partners. The objective of the present work was to identify signaling pathways affected by sclerostin in relation with its inhibitory action on osteogenic differentiation of C3H10T1/2 cells, MC3T3-E1 cells and primary osteoblasts. Sclerostin inhibited BMP2-induced osteoblast differentiation without altering SMAD1/5 phosphorylation and transcriptional activity. Moreover, sclerostin prevented Wnt3a-mediated osteoblastogenesis without affecting LRP5/6 phosphorylation or β-catenin transcriptional activity. In addition, sclerostin inhibited mineralization promoted by GSK3 inhibition, which mimics canonical Wnt signaling without activation of LRP5/6, suggesting that sclerostin can prevent osteoblast differentiation without antagonizing LRP5/6. Finally, we found that sclerostin could activate platelet-derived growth factor receptor (PDGFR) and its downstream signaling pathways PLCγ, PKC, Akt and ERK1/2. PDGFR inhibition could reverse sclerostin-mediated inhibitory activity on BMP2-induced osteoblast differentiation. Therefore, our data suggest that sclerostin can activate PDGFR signaling by itself, and this functional interaction may be involved in the negative effect of sclerostin on osteoblast differentiation.
Introduction
Characterization of two rare sclerosing bone disorders, sclerosteosis and van Buchem disease, led to the identification of the SOST gene as a negative regulator of bone formation.1,2,3,4 The SOST gene encodes sclerostin, which is synthetized and secreted postnatally by terminally differentiated osteocytes embedded in the mineralized bone matrix.5,6,7 Mice overexpressing the Sost gene display an osteoporotic phenotype with reduced osteoblast activity and bone formation but unaffected resorption.5 Conversely, Sost knockout mice exhibit a high bone mass phenotype due to increased bone formation.8 Because of its restricted expression in the adult skeleton and its inhibitory effect on bone formation, sclerostin has emerged as an attractive therapeutic target to increase bone mass and strength in osteoporotic patients. Consequently, administration of antibodies targeting sclerostin has been shown to augment bone mineral density (BMD) and bone strength in humans, through elevation of bone formation and reduction in bone resorption.9,10,11,12
During the last decade, a critical effort has been devoted to the elucidation of sclerostin mechanism of action. On the basis of the presence of a cystine-knot motif in its structure, sclerostin has been ranked in the DAN (differential screening selected gene abberative in neuroblastoma) family of secreted glycoproteins that share the ability to antagonize bone morphogenetic protein (BMP) activity.1,3 As sclerostin inhibits BMP-induced osteoblast differentiation and weakly binds BMP, it has been initially presumed to be a BMP antagonist.5,13 However, a large body of evidences has since demonstrated that sclerostin antagonizes canonical Wnt signaling by binding low-density lipoprotein receptor-related proteins (LRP) 5 and 6.14,15,16 Wnt proteins are secreted glycoproteins that bind to G protein-coupled Frizzled receptors and LRP5/6 co-receptors. This interaction results in the recruitment of Axin at the carboxy-terminal domain of LRP5/6 and inhibition of glycogen synthase kinase (GSK) 3β activity toward β-catenin. Unphosphorylated β-catenin is not degraded, accumulates in the cytoplasm, translocates to the nucleus and binds to lymphoid enhancer-binding factor/transcription factor to activate transcription of downstream target genes.17 The importance of this signaling pathway in regulating bone homeostasis has been pointed out by the identification of gain-of-function mutations of LRP5 in patients with high bone mass phenotype18,19 and loss-of-function mutations of LRP5 in patients with osteoporosis pseudoglioma syndrome.20
Interestingly, sclerostin binding to LRP5, and thus inhibition of Wnt signaling, is reduced in LRP5 gain-of-function mutants,18,21,22 further indicating that sclerostin inhibits bone formation through LRP5. In fact, it has been recently shown that sclerostin functions only partially through LRP5, as sclerostin inhibition can still increase bone mass accrual in a mouse model of osteoporosis pseudoglioma syndrome (Lrp5 deficiency).23,24 In this situation, blocking the LRP6 function with antibodies reverses bone mass gain in the cortical compartment of double Lrp5;Sost knockout mice to wild-type levels,24 indicating that sclerostin functions in part by binding to LRP6. However, remaining increased bone mass in the cancellous bone compartment in this experimental mouse model suggests that a significant part of sclerostin-mediated inhibition of bone formation is due to interactions with other binding partners. Sclerostin has been shown to interact with LRP4 in vivo.25,26 This receptor does not seem to be a component of the Wnt signaling system but has been proposed to facilitate sclerostin activity by favoring sclerostin binding to LRP5/6.25,26 In addition to LRP4/5/6, several other sclerostin-binding partners have been identified on the basis of in vitro experiments,5,27,28,29 but their respective contribution to sclerostin function remains to be addressed.
The objective of the present work was to investigate signaling pathways affected by sclerostin in relation with its inhibitory action on osteoblast differentiation in different cell culture systems.
Results
Sclerostin inhibits BMP2-induced osteoblast differentiation without affecting SMAD1/5 signaling
First, we analyzed the effects of sclerostin on BMP2-induced osteoblast differentiation of mesenchymal C3H10T1/2 cells. Recombinant sclerostin strongly inhibited BMP2-induced elevations of alkaline phosphatase (Alp) expression and ALP activity (Figures 1a and b). Sclerostin also prevented BMP2-elicited increases in expressions of osteoblast marker genes such as osterix and osteacalcin (Figure 1b). In light of these results, we next examined whether sclerostin could alter the canonical BMP signaling pathway. Sclerostin did not influence BMP2-induced SMAD transcriptional activity assessed with a BMP-responsive reporter construct, whereas noggin, a natural BMP antagonist, completely blocked it (Figure 1c). In addition, sclerostin did not affect BMP2-stimulated phosphorylation of SMAD1/5 (Figure 1d). Taken together, those data indicate that sclerostin inhibits BMP-induced osteoblast differentiation without affecting the canonical BMP signaling pathway.
Figure 1.

Sclerostin inhibits BMP2-induced osteoblast differentiation without affecting canonical BMP signaling in C3H10T1/2 cells. (a) C3H10T1/2 cells in osteogenic medium were pre-incubated with sclerostin (SOST, 5 μg ml−1) or its vehicle (Veh) for 15 min and stimulated with BMP2 (50 ng ml−1) or its vehicle (c) for 48 h before measurements of ALP activity. (b) Expression of osteoblast marker genes in the same conditions as for (a). (c) Evaluation of SMAD transcriptional activity in C3H10T1/2 cells pre-incubated with vehicle, 10 μg ml−1 sclerostin or 250 ng ml−1 noggin and stimulated with 25 ng ml−1 BMP2 or its vehicle for 16 h. (d) Western blot analyses of SMAD1/5 phosphorylation in C3H10T1/2 cells pre-treated with 10 μg ml−1 sclerostin or its vehicle and stimulated with 25 ng ml−1 BMP2 or its vehicle for different incubation times (nsp, non-specific protein). *P<0.01 for BMP2 versus control; +P<0.01 for SOST versus vehicle treatment.
Sclerostin inhibits Wnt3a-induced osteoblast differentiation without affecting canonical Wnt signaling
Next, we evaluated the effects of sclerostin on Wnt3a-induced osteoblast differentiation of C3H10T1/2 cells. Sclerostin significantly reduced the stimulation of ALP activity in response to Wnt3a (Figure 2a). Dickkopf 1 (DKK1), a natural antagonistic inhibitor of the WNT signaling pathway, exerted the same but more pronounced effect in this situation (Figure 2a). Interestingly, although DKK1 completely blocked Wnt3a-elicited β-catenin transcriptional activity tested with the TOPFlash reporter system, sclerostin did not alter it (Figure 2b). Consistent with this result, sclerostin did not affect Wnt3a-stimulated phosphorylation of LRP5/6, whereas DKK1 inhibited this response (Figure 2c). Similarly, sclerostin partially inhibited Wnt3a-induced increase in ALP activity without affecting β-catenin transcriptional activity in the pre-osteoblastic MC3T3-E1 cell line (data not shown). Therefore, our data indicate that sclerostin inhibits Wnt3a-induced osteoblast differentiation without affecting the canonical Wnt signaling pathway.
Figure 2.

Sclerostin inhibits Wnt3a-induced osteoblast differentiation without affecting canonical Wnt signaling in C3H10T1/2 cells. (a) ALP activity of C3H10T1/2 cells pre-incubated with 10 μg ml−1 sclerostin, 1 μg ml−1 dikkopf 1 (DKK1) or vehicle and stimulated with 20% control-conditioned medium or Wnt3a-conditioned medium for 3 days. (b) Evaluation of β-catenin transcriptional activity in C3H10T1/2 cells pre-incubated with 10 μg ml−1 sclerostin, 1 μg ml−1 dikkopf 1 or vehicle and stimulated with 20% control-conditioned medium or Wnt3a-conditioned medium for 24 h. (c) Western blot analyses of LRP5/6 phosphorylation in C3H10T1/2 cells stimulated with Wnt3a-conditioned medium for different incubation times; and analysis of LRP5/6 phosphorylation in C3H10T1/2 cells pre-incubated with 10 μg ml−1 sclerostin, 1 μg ml−1 dikkopf 1 or vehicle and stimulated with 200 ng ml−1 recombinant Wnt3a or its vehicle for 3 h. *P<0.01 for Wnt3a versus control; +P<0.01 for SOST or DKK1 versus vehicle treatment.
Sclerostin inhibits mineralization induced by GSK3 inhibition
Moreover, we tested the effects of sclerostin on matrix mineralization by C3H10T1/2 cells stimulated by BMP2, Wnt3a or a selective GSK3 inhibitor (SB216763). Those three osteogenic factors significantly increased matrix mineralization (Figure 3a). Coherent with its inhibitory effect on osteoblast differentiation of C3H10T1/2 cells, sclerostin inhibited Wnt3a- and BMP2-induced matrix mineralization (Figure 3a). More interestingly, sclerostin also dose-dependently inhibited matrix mineralization promoted by the selective GSK3 inhibitor (Figures 3a and b). As selective GSK3 inhibitors mimic canonical Wnt signaling without activation of LRP5/6, our findings suggest that sclerostin can prevent osteoblast differentiation without antagonizing LRP5/6, at least in C3H10T1/2 cells.
Figure 3.
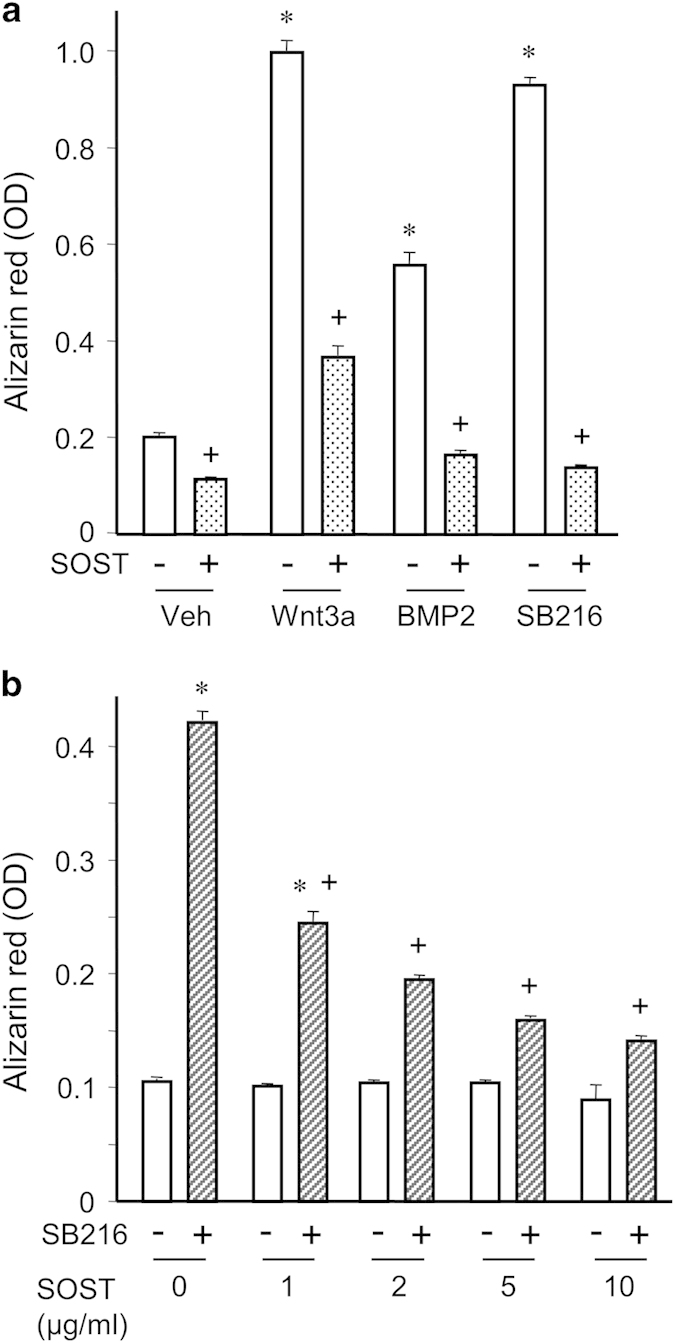
Sclerostin inhibits mineralization induced by GSK3 inhibition in C3H10T1/2 cells. (a) Quantification of matrix mineralization evaluated by Alizarin Red-S staining of C3H10T1/2 cells incubated with either vehicle, 20% Wnt3a-conditioned medium, 25 ng ml−1 BMP2 or 10 μM SB216763 (a selective GSK3 inhibitor), with or without 10 μg ml−1 sclerostin. (b) Quantification of matrix mineralization by C3H10T1/2 cells incubated with 10 μM SB216763 or its vehicle, in the presence of various doses of sclerostin. *P<0.01 for Wnt3a, BMP2 or SB216763 versus their vehicle; +P<0.01 for SOST versus its vehicle.
Sclerostin inhibits osteoblast differentiation through activation of PDGFR signaling
From the latter observation, we hypothesized that sclerostin could function through additional signaling pathways. Therefore, we investigated whether sclerostin could activate signaling pathways by itself in C3H10T1/2 and MC3T3-E1 cells. When analyzing phosphorylations of tyrosine residue, we observed that sclerostin was able to induce tyrosine phosphorylation of a protein of approximately 130 kDa in both C3H10T1/2 and MC3T3-E1 cells (Figure 4). Reblotting analyses revealed that this protein corresponded to phospholipase Cγ (PLCγ). In addition, sclerostin could activate protein kinase C (PKC), Akt and extracellular signal-regulated kinase 1/2 (ERK1/2; Figure 4) in both cell lines. As signaling pathways activated by sclerostin were very similar to those activated by platelet-derived growth factors (PDGFs), we compared signal transductions induced by sclerostin and PDGF in C3H10T1/2 and MC3T3-E1 cells. Sclerostin promoted a rapid and a sustained activation of PDGF receptors (PDGFRs) and its downstream signaling pathways (PLCγ, Akt and ERK1/2), as did PDGF-BB at low concentration (Figure 5). PDGFR inhibition with a selective inhibitor significantly reversed the inhibitory effect of sclerostin on BMP2-mediated increase in ALP activity (Figure 6), indicating that activation of PDGFR signaling is functionally involved in sclerostin-elicited inhibition of osteoblast differentiation. Finally, sclerostin significantly reduced BMP2-mediated elevation of ALP activity (Figure 7a) and could activate PDGFR signaling pathways in primary murine calvarial osteoblasts (Figure 7b).
Figure 4.

Sclerostin activates PLCγ, PKC, Akt and ERK1/2 signaling in C3H10T1/2 and MC3T3-E1 cells. (a) C3H10T1/2 and (b) MC3T3-E1 cells were exposed to 10 μg ml−1 sclerostin for various incubation times before analysis of signaling proteins by western blotting. The arrows show a protein of approximately 130 kDa, which is phosphorylated on tyrosine residues in response to sclerostin. ERK, extracellular signal-regulated kinase; PKC, protein kinase C; PLCγ, phospholipase Cγ.
Figure 5.
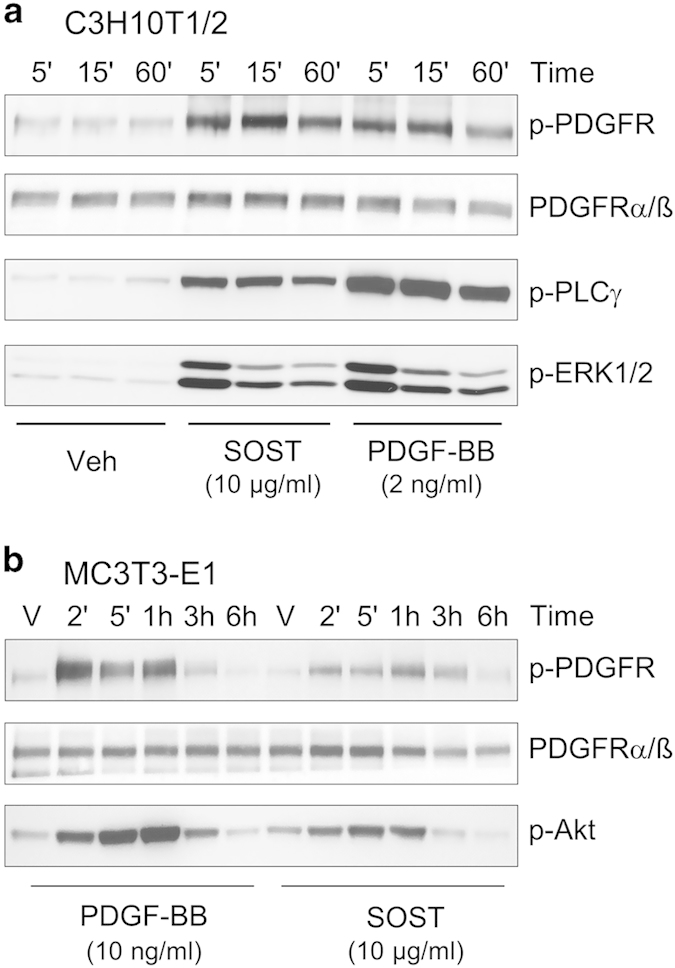
Sclerostin activates PDGFR signaling in C3H10T1/2 and MC3T3-E1 cells. (a) C3H10T1/2 and (b) MC3T3-E1 cells were treated with vehicle, 10 μg ml−1 sclerostin or 2–10 ng ml−1 PDGF-BB for various incubation times before analysis of signaling proteins by western blotting. PDGFR, platelet-derived growth factor receptor.
Figure 6.
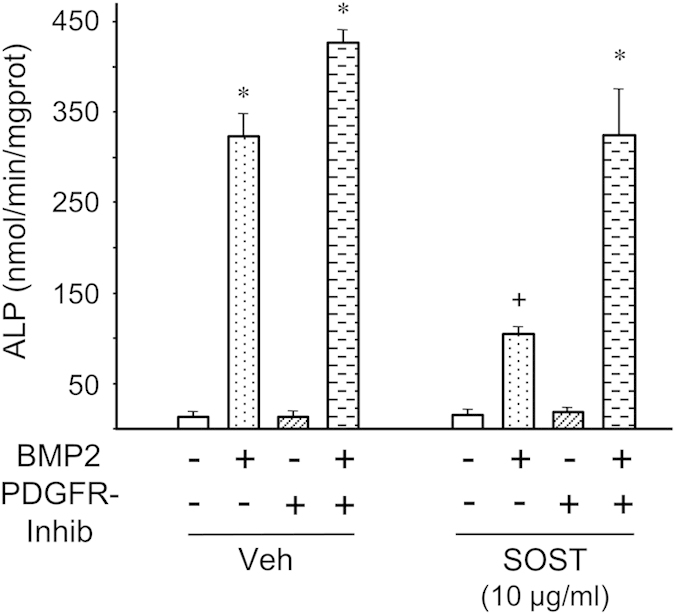
PDGFR inhibition reversed sclerostin-mediated inhibition of osteoblast differentiation. Alkaline phosphatase activity of C3H10T1/2 cells pre-incubated with 1 μM of the selective PDGFR inhibitor III or its vehicle, with sclerostin or its vehicle, and then stimulated with 25 ng ml−1 BMP2 or its vehicle for 2 days. *P<0.01 for BMP2 versus its vehicle; +P<0.01 for SOST versus its vehicle.
Figure 7.
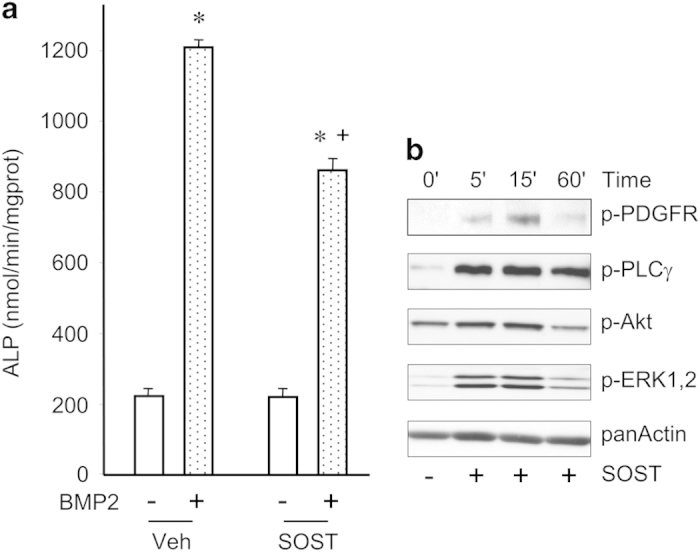
Sclerostin activates PDGFR signaling and inhibits differentiation of primary osteoblasts. (a) Primary mouse osteoblasts were pre-treated with 10 μg ml−1 sclerostin or its vehicle and stimulated or not with 100 ng ml−1 BMP2 for 48 h before evaluation of ALP activity. (b) Western blot analyses of PDGFR signaling in primary mouse osteoblasts incubated with 10 μg ml−1 sclerostin for different periods. *P< 0.01 for BMP2 versus its vehicle; +P<0.01 for SOST versus its vehicle.
Discussion
Sclerostin inhibits bone formation mostly by antagonizing LRP5/6, thus inhibiting Wnt signaling.21,22,23,24 Sclerostin also binds LRP4, which seems to facilitate sclerostin binding to LRP5/6 in this context.25,26 However, experiments with genetically modified mouse models suggest that a significant part of sclerostin-mediated inhibition of bone formation is due to interactions with other binding partners.23,24 Therefore, we aimed to identify signaling pathways affected by sclerostin in relation with its inhibitory action on osteoblast differentiation in different cell culture systems. In the investigation described here, we found that sclerostin itself could activate PDGFR signaling and that this functional interaction inhibited osteoblast differentiation.
First, sclerostin was found to inhibit BMP-induced osteoblast differentiation without affecting SMAD1/5 activation and transcriptional activity, thereby confirming that sclerostin does not function as a classical BMP antagonist.6 In addition, sclerostin prevented Wnt3a-induced osteoblast differentiation without altering LRP5/6 phosphorylation and β-catenin transcriptional activity. Recent data have highlighted the complexity of Wnt signaling with the discovery of distinct ligand binding domains in LRP5/6 receptors that recognize different classes of Wnt proteins.30 The Wnt1 class, composed of Wnt 1, 2, 6, 7a, 7b, 9a, 9b and 10b, binds β-propeller 1 of LRP5/6.30 The Wnt3a class encompasses Wnt3 and 3a and binds β-propeller 3 of LRP5/6.30 Interestingly, sclerostin has been shown to inhibit the Wnt1 class by binding to the first β-propeller region of LRP5/6 but not the Wnt3a class,24,30,31 which is coherent with our result. The fact that sclerostin could inhibit BMP2- or Wnt3a-induced osteoblast differentiation without antagonizing their respective canonical signaling suggested that it could function through another signaling pathway. This hypothesis was further strengthened by the observation that sclerostin could prevent osteoblast differentiation induced by GSK3 inhibition (which stimulates canonical Wnt signaling without activating LRP5/6).
Indeed, we demonstrated that sclerostin alone could activate PDGFR and its downstream signaling pathways PLCγ, PKC, Akt and ERK1/2 in two mesenchymal and pre-osteoblastic cell lines and in primary murine osteoblasts. In addition, activation of PDGFR signaling by sclerostin was required for the inhibitory action of sclerostin on osteoblast differentiation. A sclerostin-mediated activation of ERK1/2 in primary human osteoblasts has already been reported,32 which further supports our findings. Furthermore, we observed that sclerostin could induce a similar pattern of PDGFR signaling pathways in comparison with PDGF-BB. Intriguingly, sclerostin displays a cystine-knot domain similar to that of PDGF-BB, which is essential for its binding to PDGFR.33,34 However, it is yet to be determined whether sclerostin has the ability to interact with PDGFR. It has been proposed that sclerostin antagonizes canonical Wnt/LRP6 signaling by promoting clathrin-dependent internalization of LRP6.35 It is likely that sclerostin mediates LRP6 endocytosis through binding to a co-receptor, as it has been shown for DKK1. As PDGFR has been shown to be internalized following activation by PDGF and that PDGFR can form complex with LRP6 in other cell type,36 PDGFR could be the co-receptor involved in sclerostin-elicited endocytosis of LRP6. The fact that sclerostin could activate PDGFR signaling in primary murine osteoblasts suggests that functional interaction between sclerostin and PDGFR may occur in vivo.
Treatment with imatinib (an inhibitor of PDGFR, c-kit and c-abl) in patients with chronic myeloid leukemia and treatments with romosozumab or blosozumab (monoclonal anti-sclerostin antibodies) in post-menopausal women display similar and distinct effects on bone metabolism. Both anti-sclerostin antibodies enhanced lumbar spine, total hip and femoral neck BMDs,11,37 whereas imatinib treatment increased lumbar spine BMD but not that of total hip.38 The three treatments resulted in similar transitory increases in serum levels of bone formation markers and also promoted reduction in bone resorption markers but with different kinetics; reduction was rapid and sustained with romosozumab or blosozumab treatments, whereas it occurred after long-term therapy with imatinib.11,37,38 Finally, the three treatments led to slight decreases in serum calcium and elevations in parathyroid hormone concentrations. In contrast, slight reductions in serum phosphate and rises in 1,25-dihydroxyvitamin D3 concentration were only reported in imatinib-treated patients.11,37,38 Such indirect comparisons must be made with caution because of the differences between the two classes of drugs, the patient populations and data analysis techniques. Nevertheless, the comparable beneficial effects observed between the two types of treatment on lumbar spine BMD and bone formation, together with our in vitro observations, justify to perform a direct comparison between sclerostin and PDGFR inhibitions on bone metabolism in rodent models in future investigations.
In summary, our data demonstrate that sclerostin can activate PDGFR signaling, resulting in the inhibition of osteoblast differentiation in vitro. Thus, our results suggest that PDGFR signaling may be an important regulator of bone mass.
Materials and methods
Reagents and antibodies
Fetal calf serum (FCS), glutamine, antibiotics and trypsin/EDTA were obtained from Gibco (Life Technologies, Basel, Switzerland). Signaling inhibitors were obtained from either Calbiochem (San Diego, CA, USA) or Sigma-Aldrich (Saint Louis, MS, USA). Anti-LRP5/6 antibody was obtained from Biovision (#3801-100, Moutain View, CA, USA). Anti-p-LRP5/6 (#2568), anti-p-ERK1/2 (#9106), anti-p-Tyr (#9106), anti-p-PLCγ (#2821), anti-p-panPKC (#9371), anti-p-Akt (#4060), anti-Akt (#9272) and anti-panActin (#4968) antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). Recombinant human sclerostin, DKK1 and BMP2 were generous gifts from Novartis (Basel, Switzerland), Prostrakan (Romainville, France) and Wyeth Research (Cambridge, MA, USA), respectively. Recombinant mouse Wnt3a was purchased from R&D Systems.
Cell cultures
MC3T3-E1 and C3H10T1/2 cells were purchased from ATCC (Manassas, VA, USA). Primary mouse osteoblasts were isolated from calvaria of 3-day-old C57BL6J mice by sequential digestion with collagenase as previously described.39 MC3T3-E1 cells, C3H10T1/2 cells and primary osteoblasts were cultured in Minimum Essential Medium Eagle, alpha modifications (α-MEM) containing 10% FCS (vol/vol), 0.5% nonessential amino acids (vol/vol), 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin, at 37 °C in a humidified atmosphere of 5% CO2-95% air, and the medium was changed every 2 or 3 days. Confluent L cells and Wnt3a-producing L cells were cultured in Dulbecco's Modified Eagle Medium containing 5% FCS for 12 h for preparation of control and Wnt3a-conditioned media.
Alkaline phosphatase activity
Confluent MC3T3-E1, C3H10T1/2 and primary osteoblast cultures were switched to α-MEM containing 10% FCS, 50 μg ml−1 ascorbic acid and 10 mM β-glycerophosphate (osteogenic medium) to induce osteoblast differentiation. The day after, cultures were directly treated with the indicated agents for 2 days. ALP activity was determined as previously described.40 Essentially, cells were harvested in 0.2% Nonidet P-40 and disrupted by sonication. The homogenate was centrifuged at 1500 g for 5 min, and ALP activity was determined in the supernatant by the method of Lowry et al.41
Mineralization assays
Confluent C3H10T1/2 cells were cultured in osteogenic medium and treated with vehicle, 25 ng ml−1 BMP2, 20% (vol/vol) Wnt3a-conditioned medium or 10 μM SB216763, with or without 10 μg ml−1 sclerostin for 10 days. Matrix mineralization was analyzed by Alizarin Red-S (AR-S) staining. To quantify mineral-bound AR-S, cell cultures were destained with 10% hexadecylpyridinium chloride, and AR-S concentration was determined by measuring the absorbance at 562 nm.
Cell transfection and transient reporter assays
To analyze β-catenin transcriptional activity, subconfluent C3H10T1/2 cells were exposed to the TCF reporter plasmid (TOPFlash from Upstate Biotechnology, Lake Placid, NY, USA) and the control plasmid pRLTK expressing the Renilla luciferase (Clontech, Takara Bio Europ, France) with 225 μl PolyFect (Qiagen AG, Hombrechtikon, Switzerland) in 2 ml of α-MEM containing 10% FCS for 18 h. Agents were then added for 24 h before the determination of luciferase activity using the Promega Dual-Luciferase Assay according to the manufacturer's instructions. A similar experimental protocol was used for analyses of SMAD transcriptional activity by using a BRE luciferase reporter plasmid kindly provided by Professor Peter Ten Dijke (Leiden University, Nethertland).
Western blot analyses
Cells treated with indicated agents were rapidly frozen in liquid nitrogen after different incubation times and stored at –80 °C until used for analysis. Cell lysates were prepared by incubating cell cultures in RIPA buffer containing phosphatase and protease inhibitors at 4 °C for 30 min. Lysates were then cleared by centrifugation at 6000 g for 30 min. Lysate supernatant was diluted with an equal volume of 2-fold concentrated reducing sample buffer containing 125 mM Tris buffer (pH 6.8), 4% SDS, 20% glycerol, 0.05% bromophenol blue and 200 mM dithiothreitol. The mixture was then heated at +70 °C for 30 min and subjected to gel electrophoresis on 6% to 15% gels. After SDS–PAGE, proteins were transferred to Immobilon P membranes and immunoblotted with specific antibodies. Detection was performed using peroxidase-coupled secondary antibodies, enhanced chemiluminescence reaction and visualization by autoradiography (Amersham International, Little Chalfont, UK). Re-probed membranes were stripped according to the manufacturer's protocol.
Reverse transcription-PCR
Total RNA was extracted from C3H10T1/2 cell cultures using Tri Reagent (Molecular Research Center, Cincinnati, OH, USA) and purified using an RNeasy Mini Kit (Qiagen AG). Single-stranded cDNA was synthesized from 2 μg of total RNA using a high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Two microliters of cDNA sample was used in a 40-μl-reaction containing a Red Taq PCR reaction buffer (Sigma-Aldrich, Saint Louis, MO, USA), 1.5 mM MgCl2, 0.2 mM dATP, dCTP, dGTP, dTTP, 0.4 μM of gene-specific primers and 1 unit of Red Taq DNA Polymerase (Sigma-Aldrich). Sequences of primers used for reverse transcription-PCR analyses were previously described.42 PCR products were amplified using a program starting with 5 min of denaturation at 95 °C, followed by 30 cycles with 45 s of denaturation at 95 °C, 45 s of annealing at 58–59 °C and 30–75 s of extension at 72 °C and 5 min of final extension at 72 °C.
Statistical analysis
All experiments were carried out independently at least three times. Each independent functional analyses were performed in triplicates. Results are expressed as mean±s.e.m. of three independent experiments performed in triplicates. Comparative studies of means were analyzed using one-way ANOVA followed by a post hoc test (projected least significant difference Fisher). Differences were considered as significant when P<0.05. No statistical method was used to predetermine sample size.
Acknowledgments
We thank Sabina Troccaz and Pierre Apostolides for their expert technical assistance. This work was supported by the Swiss National Science Foundation (310030-127638).
Footnotes
The authors declare no conflict of interest.
References
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 2001; 10: 537–543. [DOI] [PubMed] [Google Scholar]
- Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 2001; 68: 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A et al. A 52- kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 2002; 110: 144–152. [DOI] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 2003; 22: 6267–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 2004; 199: 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Löwik CW et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 2005; 19: 1842–1844. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 2008; 23: 860–869. [DOI] [PubMed] [Google Scholar]
- Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 2011; 26: 19–26. [DOI] [PubMed] [Google Scholar]
- McColm J, Hu L, Womack T, Tang CC, Chiang AY. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J Bone Miner Res 2014; 29: 935–943. [DOI] [PubMed] [Google Scholar]
- McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 2014; 370: 412–420. [DOI] [PubMed] [Google Scholar]
- Papapoulos SE. Inhibition of sclerostin in the management of osteoporosis: results of a phase 2 clinical trial meet expectations. bonekey Rep 2014; 3: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem 2005; 280: 2498–2502. [DOI] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 2005; 280: 26770–26775. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 2005; 280: 19883–19887. [DOI] [PubMed] [Google Scholar]
- van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 2007; 22: 19–28. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 2006; 127: 469–480. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 2002; 346: 1513–1521. [DOI] [PubMed] [Google Scholar]
- Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 2002; 70: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001; 107: 513–523. [DOI] [PubMed] [Google Scholar]
- Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S et al. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res 2006; 21: 1738–1749. [DOI] [PubMed] [Google Scholar]
- Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem 2006; 281: 38276–38284. [DOI] [PubMed] [Google Scholar]
- Kedlaya R, Veera S, Horan DJ, Moss RE, Ayturk UM, Jacobsen CM et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med 2013; 5: 211ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MK, Kramer I, Keller H, Gooi JH, Collett C, Jenkins D et al. Reversing LRP5-dependent osteoporosis and SOST deficiency-induced sclerosing bone disorders by altering WNT signaling activity. J Bone Miner Res 2014; 29: 29–42. [DOI] [PubMed] [Google Scholar]
- Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4 a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE 2009; 4: e7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem 2011; 286: 19489–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig TA, Bhattacharya R, Mukhopadhyay D, Kumar R. Sclerostin binds and regulates the activity of cysteine-rich protein 61. Biochem Biophys Res Commun 2010; 392: 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig TA, Kumar R. Sclerostin-erbB-3 interactions: modulation of erbB-3 activity by sclerostin. Biochem Biophys Res Commun 2010; 402: 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan-Ketha H, Craig TA, Madden BJ, Robert Bergen H 3rd, Kumar R. The sclerostin-bone protein interactome. Biochem Biophys Res Commun 2012; 417: 830–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettenberg SA, Charlat O, Daley MP, Liu S, Vincent KJ, Stuart DD et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc Natl Acad Sci USA. 2010; 107: 15473–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth G, Slocombe P, Doyle C, Sweeney B, Veverka V, Le Riche K et al. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of Wnt co-receptors. J Biol Chem 2012; 287: 26464–26477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR et al. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res 2009; 24: 1434–1449. [DOI] [PubMed] [Google Scholar]
- Rey JP, Ellies DL. Wnt modulators in the biotech pipeline. Dev Dyn 2010; 239: 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem 2009; 284: 10890–10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinther M, Zhang J, Weidauer SE, Boschert V, Muth EM, Knappik A et al. Anti-Sclerostin antibody inhibits internalization of Sclerostin and Sclerostin-mediated antagonism of Wnt/LRP6 signaling. PLoS ONE 2013; 8: e62295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramati AR, Singh R, Lin A, Faramarzi S, Ye ZJ, Mane S et al. Wild-type LRP6 inhibits, whereas atherosclerosis-linked LRP6R611C increases PDGF-dependent vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA 2011; 108: 1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res 2015; 30: 216–224. [DOI] [PubMed] [Google Scholar]
- O'Sullivan S, Horne A, Wattie D, Porteous F, Callon K, Gamble G et al. Decreased bone turnover despite persistent secondary hyperparathyroidism during prolonged treatment with imatinib. J Clin Endocrinol Metab 2009; 94: 1131–1136. [DOI] [PubMed] [Google Scholar]
- Bellows CG, Aubin JE, Heersche JN, Antosz ME. Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif Tissue Int 1986; 38: 143–154. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Guicheux J, Palmer G, Miura Y, Oiso Y, Bonjour JP et al. Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone 2002; 30: 91–98. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Roberts NR, Wu ML, Hixon WS, Crawford EJ. The quantitative histochemistry of brain. II. Enzyme measurements. J Biol Chem 1954; 207: 19–37. [PubMed] [Google Scholar]
- Caverzasio J, Biver E, Thouverey C. Predominant role of PDGF receptor transactivation in Wnt3a-induced osteoblastic cell proliferation. J Bone Miner Res 2013; 28: 260–270. [DOI] [PubMed] [Google Scholar]