

Abstract
We report a biolistic technology platform for physical delivery of particle formulations of drugs or vaccines using parallel arrays of microchannels, which generate highly collimated jets of particles with high spatial resolution. Our approach allows for effective delivery of therapeutics sequentially or concurrently (in mixture) at a specified target location or treatment area. We show this new platform enables the delivery of a broad range of particles with various densities and sizes into both in vitro and ex vivo skin models. Penetration depths of ∼1 mm have been achieved following a single ejection of 200 µg high-density gold particles, as well as 13.6 µg low-density polystyrene-based particles into gelatin-based skin simulants at 70 psi inlet gas pressure. Ejection of multiple shots at one treatment site enabled deeper penetration of ∼3 mm in vitro, and delivery of a higher dose of 1 mg gold particles at similar inlet gas pressure. We demonstrate that particle penetration depths can be optimized in vitro by adjusting the inlet pressure of the carrier gas, and dosing is controlled by drug reservoirs that hold precise quantities of the payload, which can be ejected continuously or in pulses. Future investigations include comparison between continuous versus pulsatile payload deliveries. We have successfully delivered plasmid DNA (pDNA)-coated gold particles (1.15 µm diameter) into ex vivo murine and porcine skin at low inlet pressures of ∼30 psi. Integrity analysis of these pDNA-coated gold particles confirmed the preservation of full-length pDNA after each particle preparation and jetting procedures. This technology platform provides distinct capabilities to effectively deliver a broad range of particle formulations into skin with specially designed high-speed microarray ejector nozzles.
Keywords: biolistic, epidermal delivery, gene gun, intracellular delivery, injection microarrays, intradermal delivery, needle-free vaccine injection, particle ejection, particle collimation, transdermal drug delivery
Abbreviations
- pDNA
Plasmid DNA
- MEMS
MicroElectroMechanical System
- CFD
Computational Fluid Dynamics
- APC
Antigen Presenting Cells
Introduction
The development of new drug delivery methodologies focusing on precise targeting and control of the delivered payload has the potential to make significant contributions to the success of vaccines. Particle-mediated molecule delivery (i.e. biolistics) was first introduced as a promising non-invasive method for delivering payloads into cells or tissue in the mid-1980s, pioneered by Klein and colleagues, where genetic material was introduced initially into plants,1,2 later in animals,3 and finally in humans.4-6 Despite delivery challenges due to the skin's function as an effective physical barrier to pathogen entry, skin is an easily accessible, active immune organ providing an excellent site for vaccination due to its unique immunological and micro-vascular properties and extreme richness in antigen presenting cells (APCs) capable of eliciting immune responses.7 Dendritic cells (DCs), including Langerhans cells (LCs), present in the dermis and epidermis layers of skin play critical roles in antigen presentation8 and are therefore usually the target cells in powder immunization in biolistic drug delivery strategies.9 Targeting those cells could result in a significantly improved immune response to vaccine delivery compared to intramuscular (IM) routes.10 With the biolistic delivery method, formulations of a drug or vaccine are generally carried by high-density, biocompatible metal microparticles and penetrate into the target tissue after being accelerated to high speeds using pressurized gas flow.11 The delivery mechanism of particle-based delivery systems is purely physical and relies on mechanical forces to penetrate into cells or tissue and achieve the deposition of therapeutic molecules at the desired site. Various payloads have been delivered biolistically into tissue or cells in the past, including DNA,12 RNA,13 and dyes.14 Following the original design of the gene gun, efforts were devoted to improving the delivery efficiency, dose, safety, and increased immunogenicity through the development of new device designs or improvements in previous designs. A commercially available version is the Helios® gene gun (Bio-Rad, Hercules, CA) used for direct gene transfer using small plastic tubes coated with particles as drug cartridges. Upon actuation, the drug particles are swept away from the tubes by pressurized helium15 for biolistic delivery to the target site. A family of devices, based on the Contoured Shock Tube (CST) design, have been subsequently developed16 and flow characteristics were studied.17 CST-based devices have been used to study the particle penetration mechanisms into excised human skin18 and biolistic delivery to animals19 and humans in phase I clinical trials.5,20 Other capillary-based gene guns have also been reported.21
In this paper, we describe a biolistic method with distinct characteristics that uses MEMS (Micro Electro Mechanical Systems)-based devices with a multitude of micron-size ejection nozzles arrayed in parallel to provide needle-free injections. Our device creates continuous and finely tunable jets of drug particles that have been gently entrained into high-speed gas jets, enabling an explosion-free biolistic delivery system. We demonstrate that our device design offers unique characteristics by providing particle jets with high spatial resolution, allowing sequential or concurrent delivery of particles with a variety of densities, and enabling controlled penetration profiles.
Device Operating Principle and Design
Our MEMS device consists of an array of microchannels that is assembled in a prototype test cell with gas and payload inlets for device testing and performance characterization (Fig. 1A, B). As conceptually illustrated in Figure 1C, pressurized gas at the inlet causes high-speed gas jets to form inside the channels. A Venturi structure inside each channel causes rapid acceleration of the flowing gas after the constriction, which is accompanied by a localized sharp drop in pressure. Depending on the design of the Venturi structure, this pressure drop can reach sub-atmospheric levels. Figure 1D shows this concept where the pressure profiles along the center of a channel were obtained for different Venturi designs by computational fluid dynamics (CFD) simulations (Fluent).22 By placing the payload reservoir inlet next to the sub-atmospheric pressure zone, drug particles are pulled out of the reservoir and gently entrained into the high speed gas jets.23
Figure 1.

MEMS device with parallel microchannel arrays and device working principle. (A) MEMS device assembled in a prototype test cell with gas and payload inlets. (B) Scanning electron microscope (SEM) image of the fabricated device showing the parallel channels with Venturi structure and inlet of therapeutic particles. (C) Schematic illustration of operation for one channel. Pressurized gas is supplied to the inlet of the channel and accelerated after the Venturi constriction followed by particle entrainment using the mild suction generated by a brief gas pressure drop. (D) CFD simulation of pressure profile for different widths wt of the Venturi neck (wt = w, wt = w/2, and wt = w/4 where w is the channel width) along a 4 mm long channel with a cross section of 64 × 64 μm2. (E) CFD simulation of velocity profile along a 6.5 mm long channel with a cross-section of 600 × 600 μm2. Simulations in D and E used air as the pressurized gas and did not include particles.
For the entrained particles to reach the high speeds of the gas jets, a minimum channel length is required after the Venturi constriction. The required length and time to accelerate the drug particles to a given fraction of the speed of the gas jet can be obtained using the fluid mechanics drag model for a spherical particle. The equation of motion at intermediate Reynolds number is24
| (1) |
where m, v, d are the particle mass, velocity, and diameter, respectively and µ, ρf, and u are the gas viscosity, density, and speed, respectively. Using equation (1) we estimate a minimum acceleration length of a few millimeters for 50 µm diameter particles with 1000 kg/m3 density to reach 99% of the gas speed after being entrained into the channel. The exact length depends on the speed of the gas, which in turn depends on the inlet pressure and the channel cross section. Figure 1E shows CFD simulation results of the gas speed at the centerline of the channel with a cross-section of 600 × 600 μm2 confirming that the speed of the gas remains high after passing through the Venturi structure, allowing for higher final particle speeds. Therefore, the device can create continuous and finely tunable arrays of well-collimated beams of particles that may be accelerated to high velocities.
Results
In vitro studies
The two devices used in this study (‘Device A’ and ‘Device B’) have nozzle cross-sectional dimensions of 400 × 400 µm2 and 1000 × 400 µm2, respectively. Both devices had a single nozzle enabled for ejections to ensure ease of testing. Specific particle and device details are summarized in Table 1. In vitro particle penetration studies were carried out using gelatin-based targets as simulant of skin tissue. Gelatin-based materials have been demonstrated to be simple yet effective in vitro models for characterizing intradermal powder delivery.25 Such gelatin preparations are reproducible, optically clear for convenient payload visualization post-treatment, and remain stable in storage. For biolistic delivery, particles are filled in individual reservoirs that correspond to the desired dose and are introduced into the inlet port of the device through a capillary tube. Particle penetration profiles can be controlled by fine-tuning design features including nozzle size and length as well as tuning operational parameters such as ejection mode (continuous vs. pulsed), loading, and operating pressure. Payload dosing is controlled by the loading method of particles into the capillary tube and selecting the appropriate number of shots. The design of the devices enables automatic (passive) loading of particles continuously or in pulses via the vacuum generated at the payload inlet. Therefore, specified quantities of material can be ejected at a single shot and the duration of the device “on” time (i.e., the time for which the pressurized gas is flowing) can be adjusted to eject all of the material for one dose in a single shot, or to deliver it in pulses.
Table 1.
Summary of devices and particles used in in vitro penetration studies
| Devices | Nozzle cross-section | |
|---|---|---|
| Device A | 400 × 400 µm2 | |
| Device B |
1000 × 400 µm2 |
|
|
Particles |
Density (kg/m3) |
Size (µm) |
| Polystyrene-based particles | ∼1050 | 5–7 (avg. 6) |
| Tungsten | ∼19250 | avg. 1 |
| pDNA-coated gold | ∼19300 | 0.8–1.5 (avg. 1.15) |
Particle penetration studies
To adapt the developed devices for delivering payload to biological targets, the performance of the devices was characterized based on different sets of ejection conditions (i.e. inlet gas pressure, and continuous or pulsed ejections). Subsequently, the delivery of various types of particles into the gelatin-based skin simulant was assessed. After ejection of the particles, the maximum depth of penetration into the gel was quantified by microscopy and image analysis. Figure 2 shows the penetration depth of different particles with various densities from ∼1050 kg/m3 to ∼19300 kg/m3 (Table 1). Gold and tungsten were used as higher density particles in these experiments. It is important to note that tungsten particles possess a density that closely matches that of gold and thus were used as mock particles solely for device characterization purposes; thus they were not used in biological studies.
Figure 2.

Cross-sectional images of particles penetration profiles after ejection into gelatin at 70 psi inlet pressure. Device A (400 × 400 µm2 channel cross-section) was used for ejections in (A-D, G) and Device B (1000 × 400 µm2 channel cross-section) was used for ejection in (E) where both devices had a single nozzle enabled for ejections. The ejection dose was ∼13.6 µg for low-density particles shown in (A), 200 µg for high-density particles shown in B and C, ∼215 µg for mixture of particles shown in (D and E) and a total of 1mg for multiple ejections shown in G. (A) Penetration of low-density polystyrene-based particles with density of ∼1050 kg/m3. (B) Penetration of tungsten particles with density of ∼19250 kg/m3. (C) Penetration of gold particles with density of ∼19300 kg/m3 that were coated with pDNA at 10 µg DNA in 1 mg of gold. (D and E) Penetration of a mixture of low and high density particles. (F) Maximum penetration of particles in a-e (n = 3). The error bars represent one standard deviation from measured values. (G) Penetration of pDNA-coated gold particles after 5 sequential ejections for 1 mg dose delivery.
The devices ejected 200 µg of high-density particles and ∼13.6 µg of low density particles from 40 nl reservoirs when operated at 70 psi with a single shot using a single nozzle in less than 1 s. All types of particles show more than 1 mm penetration into gelatin following a single ejection of the dose (Figure 2A, B, and C). Particle penetration reached ∼3 mm after ejection in 5 pulses of 200 µg dose each, for a total of 1 mg dosing at a similar gas pressure of 70 psi (a representative penetration profile is shown in Fig. 2G).
Controlling the penetration depth
It is desirable to tune the penetration depth of particles for targeting specific cells or layers of tissue. Particle penetration depths were assessed after performing single ejections into gelatin-based targets while the device was operated using various inlet gas pressures. Figure 3A demonstrates the cross-sectional images of the penetration depth profiles after the ejection of 200 µg tungsten particles at 45 psi, 65 psi, and 70 psi inlet gas pressures. Repeatable ejections and penetration depths were achieved with controlled payload quantities. Also, the footprints of ejections were smaller than 1 mm2 for all the conditions used and may be tuned in different designs. The results confirm that penetration depth of particles in gel can be tuned by adjusting the inlet gas pressure. Similar results were observed with other types of particles (data not shown). We demonstrated earlier (Fig. 2G) that multiple ejections at one location increase the penetration depth and total dose of particles in the gel.
Figure 3.

Operating pressure effect on particle penetration after ejection into gelatin. (A) Representative cross-sectional images of tungsten particles penetration into gelatin samples using Device A at various operating pressures. (B) Maximum penetration of tungsten particles (200 µg dose) increases while the inlet pressure increases following single ejection (n = 3). (C) Device ejection efficiency defined as the extracted pDNA-coated gold particles from the gel after ejection of defined quantities of particles in 5 consecutive pulses at 70 psi (n = 6). The error bars represent one standard deviation from measured values.
Ejection efficiency
Estimating the ejection efficiencies of the devices enables better dose planning for maintaining the therapeutic drug concentration above minimum efficacy levels. Plasmid DNA (pDNA)-coated gold particles at a concentration of 10 µg plasmid per 1 mg gold were loaded into the devices in precise quantities and ejected in 5 consecutive pulses into the gelatin-based targets at an operating pressure of 70 psi. Particles were extracted from the gel and the ejection efficiency—defined as the ratio of the amount of gold particles harvested from the gel post-ejection (not depth specific) to the total amount initially loaded to the device—was derived. Efficiencies of ejections were 60% and 56% for Device A and B, respectively (Fig. 3C). The losses during this process may be associated with several factors, such as non-ideal recovery of particles from the treated gel samples or particles not being captured in the target. To achieve therapeutic effect thresholds of vaccines or therapies, the ejection efficiencies of the devices should be factored in the loading quantity of payload to achieve required dosing.
Plasmid DNA integrity
To test the effect of particle preparation, loading, and jetting on the integrity of pDNA coated on gold particles, samples from each step of the process were collected, pDNA was eluted in TE buffer and run on a 1% agarose gel. With the inlet pressure set at 70 psi, 5 shots of pDNA-coated gold particles (approximately 1 mg total) were ejected and captured in a 15 ml tube containing 0.5 ml glycerol. Figure 4 shows the agarose gel analysis data in various steps of particle preparation and jetting using both devices A and B. The results of agarose gel electrophoresis show that full-length pDNA was preserved at each step of particle preparation. This confirms that high speed ejection of particles does not affect the integrity of pDNA. However, the analysis shows a change in the ratio of the supercoiled conformation to all conformations present in gWiz plasmid (i.e., relaxed circular, linear, and supercoiled) during particle processing (i.e. suspending and dispersing before loading) and jetting.
Figure 4.
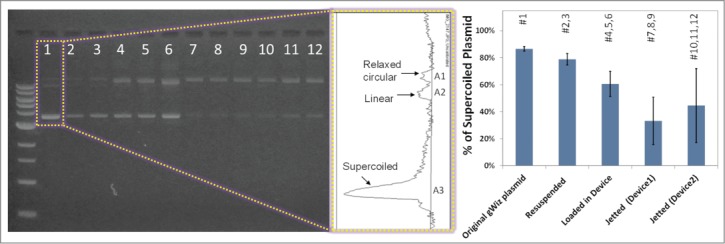
The effect of particle preparation, processing before loading, and jetting on the integrity of pDNA coated on gold particles. The graph shows the ratio of the supercoiled conformation to all forms present in the original gWiz plasmid.
Ex vivo studies
Delivery of pDNA-coated gold particles into ex vivo skin
For further clinical applications it is important to demonstrate the feasibility of our devices for biolistic delivery of therapeutics into tissues without any detectable tissue damage. Therefore, device performance was assessed on ex vivo skin models that included both epidermis and dermis layers. Both murine and porcine skin samples were stretched to replicate live skin tension in order to better mimic in vivo conditions in our study. Prior work has shown that hydration in ex vivo skin models influences the penetration depth of particles following the biolistic ejections.26 Therefore, to maintain consistency in ex vivo skin hydration, the freshly excised skin tissues were exposed to phosphate buffered saline (PBS) soaked fabric before each ejection. We ejected pDNA-coated gold particles onto freshly excised murine and porcine skin tissues mounted over a soft gel substrate. Studying penetration of particles using ex vivo mouse skin models facilitates comparison of performance with prior biolistic ejectors as murine animal models have been widely used for immunological studies27-29 and porcine skin has often been used in jet injecting penetration studies30 due to its similarity to human skin structure.31 Figure 5A demonstrates a schematic diagram of the experimental setup where Device B was operated at low pressures (less than 30 psi) to eject pDNA-coated gold particles with approximately 10 µg pDNA per 1 mg gold. Figure 5B and C are histology images of the mouse skin tissue stained with hematoxylin and eosin (H&E) and visualized using bright and dark field microscopy revealing particles penetrating more than 100 µm into the skin. Ejection in porcine skin (Fig. 5D) shows the potential for deep particle penetration even under low device operating pressure.
Figure 5.
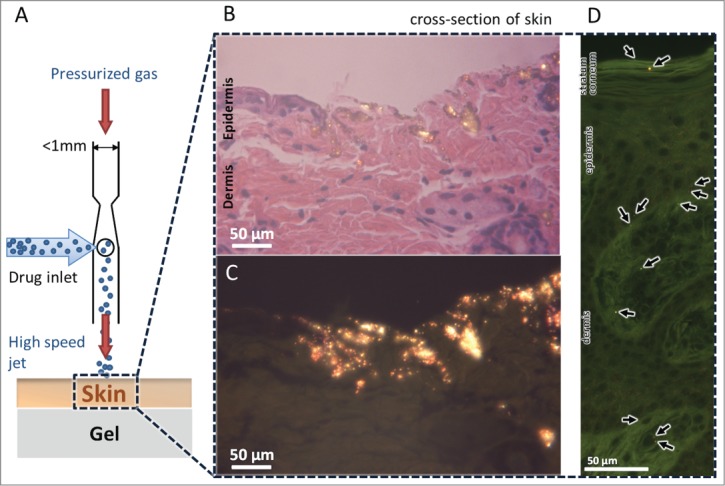
Delivery of pDNA-coated gold particles into the ex vivo skin models. (A) Schematic diagram of a single nozzle of a device operating to deliver pDNA-coated gold particles into the freshly excised skin placed over a gel as a soft substrate. Schematic is not to scale. (B) Histology of mouse skin tissue by hematoxylin and eosin (H&E) staining after ejected with gold particles, visualized by bright field microscopy. (C) Dark field microscopy of histology sample in B reveals the glowing gold particles penetrated into the skin. (D) Histology of pig skin tissue by H&E staining after bombardment visualized by dark field microscopy.
Discussion
This paper describes a biolistic delivery platform with distinct capabilities and investigates particle penetration using in vitro skin simulant and ex vivo skin models.
As depicted in Figure 2, ejection of drug particles using our devices generates highly collimated focused beams of particles (particle jets) impinging into the substrate, demonstrating very different penetration profiles from those using previously developed biolistic ejectors featuring diverging particle trajectories and shock-tube based approaches.18,32,33 The collimated particle delivery method is advantageous and opens up new therapeutic strategies as the particle landing locations may be precisely and repeatedly defined. For example, as shown in Figure 2D and E, co-delivery of various payloads is possible at one location, sequentially or as a mixture, with micrometer precision. Therefore, because the particle landing location is accessible repeatedly during multiple sequential ejections via each device nozzle, these devices may enable the mixing and reaction of compounds at the delivery site that could potentially simplify drug formulation processes. The collimated particle delivery technology may be especially advantageous for intracellular delivery, applicable in the transdermal delivery of nucleic acid-based therapies, or in applications where microscale spatial precision is required to accurately target functional particles at desired locations and depths in the skin.
It is important to note that all the ejections reported here as shown in Figure 2 have used only a single nozzle of the multi-nozzle devices. Depending on the application, an entire array of nozzles can be used in parallel (Fig. 1) to achieve hundreds of simultaneous ejections in less than one second at a desired spatial resolution down to tens of micrometers, allowing uniform distribution of particles with defined profiles at defined depths with defined distances from each other, capabilities different from previous single nozzle ejectors. Moreover, for large area delivery applications, our device architecture (with micron size channels) could enable configurations where target skin is being scanned/rastered automatically while the payload is ejected on-demand—very similar to printing ink on paper where printer head is moving along the paper and marking specified patterns. This device configuration will be investigated in future for therapeutic delivery applications.
Penetration results shown in Figure 2A demonstrate significant potential for the delivery of low-density particles at relevant depths and hence ‘pure’ drug particles. As such, the need for high-density carriers (i.e., gold particles) may be eliminated. This is an important advantage of the developed ballistic drug delivery approach as the delivery dosing efficiency will dramatically increase, which could eliminate the potential toxicities and known inefficiencies associated with using non-therapeutic carrier particles. We speculate that such focused beams of particles may be the enabler for pure drug particle delivery at depths approaching those achieved by high-density gold carrier particles. Further studies are currently being planned to verify this effect in vivo.
The two device designs used in this work led to distinct capabilities that could be tailored to different applications. As shown in Figure 3B, Device A provides superior penetration depths into gelatin-based samples than Device B at 45 and 70 psi. Better collimation provided with Device A, having 2.5 times smaller cross-sectional area than Device B as well as more symmetrical ejection nozzle (i.e. equal cross-sectional height and width) may be responsible for better penetration depths achieved by Device A at those pressures. The optimal penetration performance of Device B was achieved at 60 psi operating pressure in these in vitro tests. Thus, Device B is likely more suitable for penetrations into softer tissues.
Depending on the application requirements, smaller and larger dosing is possible using our delivery technology. Dosing is determined by the quantities loaded and can be regulated by the size of the reservoir. Dose delivery can be controlled in 2 ways: a) by the individual reservoir size and selecting the number of ejection pulses, b) by the duration of a continuous ejection from a large reservoir. The quantity of payload entrained per pulse should be optimized to avoid overloading the ejection nozzle, which may negatively affect particle velocity inside the nozzle.
Full-length pDNA was preserved during ejection of pDNA-coated gold particles from the device, although the ratio of the supercoiled conformation to all conformations was reduced. It is not evident if this change in conformation will have a direct effect in eliciting an immune response using these particles. Although prior studies have suggested that optimal transfection and immnogenicity are achieved by injecting the supercoiled conformation of pDNA, the relaxed circle and linear plasmids have also been shown to be immunogenic as DNA vaccines in animals.34 This effect requires further investigations in the future.
To demonstrate the potential for the developed biolistic delivery system to be further developed into a powerful in vivo-relevant therapeutic tool, the ex vivo skin models were setup in an attempt to mimic in vivo conditions and were jetted with gold carrier particles coated with pDNA. The ejections of these particles into the ex vivo skin tissues were performed in low pressures (<30 psi) and showed penetration depths of more than 100 µm in mouse skin. The design of our devices allows for operation in much higher inlet gas pressures (>30 psi) that results in a substantial increase in the velocity of entrained particles by the accelerated carrier medium after the Venturi structure. This effect has been demonstrated in vitro (Fig. 3) where particle penetration depths were tuned by increasing the operating inlet pressures. In human skin applications, this device characteristic may enable targeting drug particles into specific cells or tissue on-demand, an effect that will be studied in further detail in the future. Local skin reactions to a particle-based immunization have been reported in a phase 1 clinical study using CST-based particle delivery devices operated with a nominal gas pressure of ∼650 psi.20 In that study, biolistic ejections were found safe with mild self-limiting local skin reactions that resolved after 14–28 days where pain, tingling, or paresthesia were reported infrequent. Another clinical study reported that every patient felt less pain after receiving biolistic ejection with their devices (gas pressure at ∼464 psi) compared to traditional intramuscular injection.35 Such information for the biolistic technology described in this paper will be useful in human skin applications. It is likely that skin mechanoreceptors, especially low threshold skin's Merkel's discs to play a role in sensing the applied pressure during ejection and skin's Pacinian corpuscles to produce sensation of vibration or tickle for transient disturbances at frequencies in the range of 250–350 Hz which may happen during biolistic ejections.36
Data presented in this work suggests that the developed biolistic technology platform may create new therapeutic strategies and paradigms for effective delivery of a broad range of particle formulations of drugs or vaccines into human skin. The collimated jets demonstrated in this work may open a new therapeutic route for highly localized non-invasive treatments and immunizations that can be adapted to a number of therapeutic applications as the devices and their operating parameters can be designed and optimized to achieve specific delivery goals such as penetration depths for specific targets. Possible clinical applications for this technology may be in the areas of mass immunization performing many ejections from a large reservoir of therapeutics, delivery of vaccines for infectious disease including pandemics, cancer immunotherapy for targeting antigen presenting cells (APCs), melanoma skin cancer treatment, delivery of immunosuppressive treatments, and co-delivery of antigens and immunomodulatory agents antagonizing the immunosuppressive environment of different diseases to achieve effective immune responses. Because this technology allows the delivery of a wide range of particles with various densities, it may also find applications in delivery of cosmetic products into the skin or mucosal tissue, which benefit from precise targeting and controlled penetration depths, as well as delivery of nutritional/nourishing substances, long-term sun-screen, medications for wound healing or other dermatological conditions.
Several other aspects of this technology remain to be explored in future in vivo studies. One area involves the induction of antibody responses or reporter gene expression in animal models following biolistic delivery using our microarrayed devices and comparing the responses when active materials are in the form of high and low density particles. Given the spatial precision capability of our platform, another area of future investigation involves the in vivo exploration of co-delivery and therefore co-location of various types of therapeutic particles at the delivery site to enhance or modulate the responses. Additionally, comparison between continuous versus pulsatile payload delivery and the delineation of specific applications for which pulsatile or continuous ejection is better suited are areas to be explored in future.
Methods and Materials
Device fabrication and assembly
The fabrication of the devices involved standard MEMS processing techniques including photolithography for patterning the channels and Venturi structures, followed by deep-reactive ion etching (DRIE) for creating the desired structures in silicon. Similarly, the payload inlet holes were created by backside alignment, photolithographic patterning, and DRIE etching of the holes in silicon. The channels were then sealed by glass cover chips using anodic bonding. Gas inlet ports were created by drilling into the cover glass. The overall dimensions of the device and the number of channels in each device may be designed based on the application requirements, and can be fine-tuned as needed. The typical overall dimensions of individual devices were 10 × 10 × 0.5 mm3. The devices were then assembled into a prototype test cell (Fig. 1A) to provide an interface between the device inlets and controlled pressurized gas as well as individual drug reservoirs through a capillary tube that was assembled onto the device payload inlet. The drug particles were supplied from drug reservoirs to the end of the capillary tube. Payload particles were allocated into drug reservoirs using a squeegee process.
Production of pDNA-coated particles
pDNA-coated gold particles were prepared as described in the Bio-Rad Helios Gene Gun System Instruction Manual except that 100% ethanol was used in place of polyvinylpyrrolidone (PVP).37 The goal was to obtain well-separated particles and therefore PVP was omitted because it acts as an adhesive during the gene gun cartridge preparation process.37 To prepare DNA-coated gold particles at a concentration of 10 µg DNA/mg gold, 50 mg of 0.8–1.5 µm (average 1.15 µm) diameter spherical gold particles (Alfa Aesar Cat. No. 39817) were mixed with 0.5 ml 0.05 M spermidine (Sigma Aldrich Cat. No. 85558). The mix was sonicated for 5 min in an ultrasonic bath. While vortexing the mixture, 500 µg of gWiz-SEAP pDNA diluted in TE buffer (Sigma Aldrich Cat. No. T8280) to 1 mg/ml from the stock solution (Aldevron Cat. No. 5005) was added dropwise. The gold, spermidine, and DNA mix was vortexed and 0.5 ml 1 M CaCl2 (Sigma Aldrich Cat. No. C4901) was added dropwise to precipitate the DNA onto the gold particles. After 15 minutes incubation at room temperature, the DNA-coated gold particles were spun down by brief centrifugation at 5,000 RCF, liquid was aspirated, and the gold was washed 3 times with 1 ml cold 100% ethanol (Sigma Aldrich Cat. No. E7023) each time. The gold particles were suspended in 1 ml 100% ethanol, transferred to a 75 × 50 mm glass slide and dried at room temperature in a fume hood. Aggregates of gold particles were mechanically separated before loading the dispersed gold particles into the reservoirs from where they were loaded into the device via capillary.
Preparation of gelatin-based skin simulant
Skin simulant samples were prepared using a gelatin/glycerol mixture (weight ratio 3.5/6.5) using gelatin from porcine skin for microbiology with a Bloom strength of 180 (Sigma Aldrich Cat. No. 48722) and glycerol for molecular biology (Fisher Scientific Cat. No. BP2291). These methods follow those described previously in the literature.25 Deionized water and glycerol were independently warmed to ∼55°C. Gelatin powder was added to the warmed deionized water to produce a 10% (w/w) solution. Appropriate amounts of the 10% gelatin solution and glycerol were combined to achieve the 3.5/6.5 gel/gly weight ratio. The mixture was stirred and degassed for 5 min at ∼55°C, and subsequently poured into Petri dishes to cool and dry in a chemical hood for 72 hours at ambient conditions. Each gelatin preparation was then peeled, flipped, and allowed to dry for another day in the chemical hood. Circular samples (∼12 mm diameter and ∼5 mm thickness) of the prepared skin simulant were then mounted onto glass microscope slides for each jetting experiment.
Harvesting of animal skin
Animal subjects and veterinary expertise were provided by PMI - Preclinical Medevice Innovations. The method for harvesting skin samples follows those described previously in literature.30 Skin samples from porcine and murine subjects were collected within 2 hours post-euthanasia. Excised porcine skin was obtained from the abdominal skin area lateral to the mammary glands of a given Yorkshire pig (weight range 80 – 90 kg). Murine skin was obtained from dorsal and ventral areas of balb/c mice (approx. 6 weeks old). In each case, depilatory cream (Nair) was applied to the described areas for 10–15 min followed by a thorough rinsing and cleaning with warm, damp wipes to remove all residual depilatory cream. Sharp or abrasive tools were avoided during this process in order to limit damage to the skin. The importance of stretching skin samples to original dimensions in ex vivo ejection studies was highlighted previously.38 Here, to replicate live skin tension in both murine and porcine skin samples a surgical marking pen was used to trace a pre-measured template directly on the skin so that the samples could be stretched to the appropriate original dimensions during subsequent jetting experiments. Skin was cut and underlying fat, if present, was removed using a surgical scalpel. Skin pieces were rinsed and moistened with phosphate buffered saline and stored in a cool environment to be used for ejection experiments within a few hours.
Jetting of payload particles
Three different types of payload particles were ejected from devices: pDNA-coated gold particles (prepared in-house), 1 µm average diameter tungsten particles (Noah Technologies Corporation Cat. No. 17128), and polystyrene-based particles from a custom formulation obtained from XRCC (Xerox Research Center of Canada). Devices were oriented such that particles would be jetted along a horizontal axis toward a target substrate mounted perpendicular to the jetting direction, approximately 1 mm from the nozzle exit. Either the prepared skin simulant or harvested skin served as the target substrate. Controlled duration of pressurized helium or air at selected pressures was introduced into the device to entrain and propel the payload toward the target substrate.
Characterization of the depth of particle penetration in gel
To visualize a cross-section of the trajectory of the penetrated payload, a surgical blade (Feather® Safeshield™ Disposable no. 10 Scalpel, VWR Cat. No. 102097-798) was used to carefully slice the gelatin sample in a direction parallel to the particle penetration pathways approximately 2 mm away from the jetted site by positioning the blade perpendicular to the impingement surface. This exposed surface of the cross-section was mounted onto a coverslip to maintain a flat plane for bright field microscopy using an Olympus BH-2 microscope with 5× and 10× NeoSPlan objectives. All images were processed using ImageJ® software to quantify the linear distance from the impingement surface to the deepest penetrated particles.
Histological processing and analysis of skin samples
After particle ejection into the excised skin samples, the area was marked, gently rinsed with phosphate buffered saline to remove any excess particles on the skin surface, and cut using 4 mm diameter biopsy punches. Each biopsy sample was fixed in neutral buffered 10% formalin (Fisher Scientific Cat. No. 23305510) at room temperature for at least 48 hours. Subsequent paraffin-embedding, sectioning, and hematoxylin and eosin staining were performed by Histo-Tec Laboratory (Hayward, CA). Each biopsy sample was sectioned to reveal cross-sections of the payload penetration trajectory such that one prepared histology slide would capture the tissue penetration from the impingement surface to the deepest deposited particles. Prepared histology slides were analyzed using bright and dark field microscopy using a Nikon Eclipse LV100 microscope with a Nikon LU Plan 50× ELWD objective.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Meng Lean, Ben Hsieh, Fred Endicott, Philip Floyd, Jaan Noolandi, David Biegelsen, Raj Apte, and G.A. Neville Connell for early discussions and contributions.
References
- 1.Klein TM, Wolf ED, Wu R, Sanford JC. High-velocity microprojectiles for delivering nucleic acids into living cells. Nature 1987; 327:70-3; http://dx.doi.org/ 10.1038/327070a0 [DOI] [PubMed] [Google Scholar]
- 2.Sanford JC, Klein TM, Wolf ED, Allen N. Delivery of substances into cells and tissues using a particle bombardment process. Particul Sci Technol 1987; 5:27-37; http://dx.doi.org/ 10.1080/02726358708904533 [DOI] [Google Scholar]
- 3.Williams RS, Johnston SA, Riedy M, DeVit MJ, McElligott SG, Sanford JC. Introduction of foreign genes into tissues of living mice by DNA-coated microprojectiles. Proc Natl Acad Sci U S A 1991; 88:2726-30; PMID:2011582; http://dx.doi.org/ 10.1073/pnas.88.7.2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy MJ, Wu MS, Barr LJ, Fuller JT, Tussey LG, Speller S, Culp J, Burkholder JK, Swain WF, Dixon RM, et al.. Induction of antigen-specific CD8+ T cells, T helper cells, and protective levels of antibody in humans by particle-mediated administration of a hepatitis B virus DNA vaccine. Vaccine 2000; 19:764-78; PMID:11115698; http://dx.doi.org/ 10.1016/S0264-410X(00)00302-9 [DOI] [PubMed] [Google Scholar]
- 5.Drape RJ, Macklin MD, Barr LJ, Jones S, Haynes JR, Dean HJ. Epidermal DNA vaccine for influenza is immunogenic in humans. Vaccine 2006; 24:4475-81; PMID:16150518; http://dx.doi.org/ 10.1016/j.vaccine.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 6.Dean HJ, Fuller DH, Yager EJ. Prospects for developing an effective particle-mediated DNA vaccine against influenza. Expert Rev Vaccines 2009; 8:1205-20; PMID:19722894; http://dx.doi.org/ 10.1586/erv.09.82 [DOI] [PubMed] [Google Scholar]
- 7.Salmon JK, Armstrong CA, Ansel JC. The skin as an immune organ. West J Med 1994; 160:146-52; PMID:8160465 [PMC free article] [PubMed] [Google Scholar]
- 8.Guy B, Nicolas J-F. Intradermal, epidermal and transcutaneous vaccination: from immunology to clinical practice. Expert Rev Vaccines 2008; 7:1201-14; PMID:18844594; http://dx.doi.org/ 10.1586/14760584.7.8.1201 [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Payne LG. Targeting epidermal Langerhans cells by epidermal powder immunization. Cell Res 2002; 12:97-104; PMID:12118944; http://dx.doi.org/ 10.1038/sj.cr.7290115 [DOI] [PubMed] [Google Scholar]
- 10.Kersten G, Hirschberg H. Needle-free vaccine delivery. Expert Opin Drug Deliv 2007; 4:459-74; PMID:17880271; http://dx.doi.org/ 10.1517/17425247.4.5.459 [DOI] [PubMed] [Google Scholar]
- 11.Haynes JR. Particle-mediated DNA vaccine delivery to the skin. Expert Opin Biol Ther 2004; 4:889-900; PMID:15174971; http://dx.doi.org/ 10.1517/14712598.4.6.889 [DOI] [PubMed] [Google Scholar]
- 12.Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci U S A 1993; 90:11478-82; PMID:8265577; http://dx.doi.org/ 10.1073/pnas.90.24.11478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu P, Ziegelhoffer P, Sun J, Yang N. Gene gun delivery of mRNA in situ results in efficient transgene expression and genetic immunization. Gene Ther 1996; 3:262-8; PMID:8646558 [PubMed] [Google Scholar]
- 14.O'Brien JA, Lummis SCR. Diolistics: incorporating fluorescent dyes into biological samples using a gene gun. Trends Biotechnol 2007; 25:530-4; http://dx.doi.org/ 10.1016/j.tibtech.2007.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin MTS, Pulkkinen L, Uitto J, Yoon K. The gene gun: current applications in cutaneous gene therapy. Int J Dermatol 2000; 39:161-70; PMID:10759952; http://dx.doi.org/ 10.1046/j.1365-4362.2000.00925.x [DOI] [PubMed] [Google Scholar]
- 16.Kendall MAF. The delivery of particulate vaccines and drugs to human skin with a practical, hand-held shock tube-based system. Shock Waves 2002; 12:23-30; http://dx.doi.org/ 10.1007/s001930200126 [DOI] [Google Scholar]
- 17.Kendall MAF, Quinlan NJ, Thorpe SJ, Ainsworth RW, Bellhouse BJ. Measurements of the gas and particle flow within a converging-diverging nozzle for high speed powdered vaccine and drug delivery. Exp Fluids 2004; 37:128-36; http://dx.doi.org/ 10.1007/s00348-004-0792-4 [DOI] [Google Scholar]
- 18.Kendall M, Mitchell T, Wrighton-Smith P. Intradermal ballistic delivery of micro-particles into excised human skin for pharmaceutical applications. J Biomech 2004; 37:1733-41; PMID:15388316; http://dx.doi.org/ 10.1016/j.jbiomech.2004.01.032 [DOI] [PubMed] [Google Scholar]
- 19.Loudon PT, Yager EJ, Lynch DT, Narendran A, Stagnar C, Franchini AM, Fuller JT, White PA, Nyuandi J, Wiley CA, et al.. GM-CSF increases mucosal and systemic immunogenicity of an H1N1 influenza DNA vaccine administered into the epidermis of non-human primates. PLoS One 2010; 5:e11021; PMID:20544035; http://dx.doi.org/ 10.1371/journal.pone.0011021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean HJ, Chen D. Epidermal powder immunization against influenza. Vaccine 2004; 23:681-6; PMID:15542190; http://dx.doi.org/ 10.1016/j.vaccine.2004.06.041 [DOI] [PubMed] [Google Scholar]
- 21.Rinberg D, Simonnet C, Groisman A. Pneumatic capillary gun for ballistic delivery of microparticles. Appl Phys Lett 2005; 87:014103; http://dx.doi.org/ 10.1063/1.1951044 [DOI] [Google Scholar]
- 22.Volkel AR, Anderson GB, Endicott FJ, Chow EM, Pattekar AV. Ballistic aerosol marking. The 17th International Conference on Solid-State Sensors, Actuators and Microsystems (TRANSDUCERS & EUROSENSORS XXVII) 2013:2385-8 [Google Scholar]
- 23.Hsieh HB, Pattekar AV, Uhland S, Völkel AR, Recht M, Linn F, Anderson GB, Chow EM. Development of novel arrayed microjet devices for transdermal drug administration. 40th Annual Meeting & Exposition of the Controlled Release Society. Hawaii, USA, 2013. [Google Scholar]
- 24.Zhu C, Fan L-S. Multiphase Flow: Gas/Solid. : Johnson RW, ed. The handbook of fluid dynamics. 1 ed., 18: CRC Press; 1998:1-48. [Google Scholar]
- 25.Deng Y, Winter G, Myschik J. Preparation and validation of a skin model for the evaluation of intradermal powder injection devices. Eur J Pharm Biopharm 2012; 81:360-8; PMID:22484250; http://dx.doi.org/ 10.1016/j.ejpb.2012.03.008 [DOI] [PubMed] [Google Scholar]
- 26.Kendall M, Rishworth S, Carter F, Mitchell T. Effects of relative humidity and ambient temperature on the ballistic delivery of micro-particles to excised porcine skin. J Invest Dermatol 2004; 122:739-46; PMID:15086561; http://dx.doi.org/ 10.1111/j.0022-202X.2004.22320.x [DOI] [PubMed] [Google Scholar]
- 27.Cu Y, Broderick K, Banerjee K, Hickman J, Otten G, Barnett S, Kichaev G, Sardesai N, Ulmer J, Geall A. Enhanced delivery and potency of self-amplifying mRNA vaccines by electroporation in situ. Vaccines 2013; 1:367-83; http://dx.doi.org/ 10.3390/vaccines1030367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Kask AS, Crichton ML, McNeilly C, Yukiko S, Dong L, Marshak JO, Jarrahian C, Fernando GJP, Chen D, et al.. Improved DNA vaccination by skin-targeted delivery using dry-coated densely-packed microprojection arrays. J Control Release 2010; 148:327-33; PMID:20850487; http://dx.doi.org/ 10.1016/j.jconrel.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 29.Combadière B, Mahé B. Particle-based vaccines for transcutaneous vaccination. Comp Immunol Microbiol Infect Dis 2008; 31:293-315; http://dx.doi.org/ 10.1016/j.cimid.2007.07.015 [DOI] [PubMed] [Google Scholar]
- 30.Schramm J, Mitragotri S. Transdermal drug delivery by jet injectors: energetics of jet formation and penetration. Pharm Res 2002; 19:1673-9; PMID:12458673; http://dx.doi.org/ 10.1023/A:1020753329492 [DOI] [PubMed] [Google Scholar]
- 31.Vardaxis NJ, Brans TA, Boon ME, Kreis RW, Marres LM. Confocal laser scanning microscopy of porcine skin: implications for human wound healing studies. J Anat 1997; 190(Pt 4):601-11; PMID:9183682; http://dx.doi.org/ 10.1046/j.1469-7580.1997.19040601.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoecklinger A, Grieshuber I, Scheiblhofer S, Weiss R, Ritter U, Kissenpfennig A, Malissen B, Romani N, Koch F, Ferreira F, et al.. Epidermal langerhans cells are dispensable for humoral and cell-mediated immunity elicited by gene gun immunization. J Immunol 2007; 179:886-93; PMID:17617579; http://dx.doi.org/ 10.4049/jimmunol.179.2.886 [DOI] [PubMed] [Google Scholar]
- 33.Steele KE, Stabler K, VanderZanden L. Cutaneous DNA vaccination against Ebola virus by particle bombardment: histopathology and alteration of CD3-positive dendritic epidermal cells. Vet Pathol 2001; 38:203-15; PMID:11280377; http://dx.doi.org/ 10.1354/vp.38-2-203 [DOI] [PubMed] [Google Scholar]
- 34.Liu MA, Ulmer JB. Human Clinical Trials of Plasmid DNA Vaccines. In: Hall JC, Dunlap JC, Friedmann T, Heyningen Vv, eds. Advances in Genetics. 1 ed., 55: Academic Press; 2005:25-40. [DOI] [PubMed] [Google Scholar]
- 35.Brouillette M, Doré M, Hébert C, Spooner MF, Marchand S, Côté J, Gobeil F, Rivest M, Lafrance M, Talbot BG, et al.. A new biolistic intradermal injector. Shock Waves 2013:1-13 [Google Scholar]
- 36.In: Purves D, Augustine GJ, Fitzpatrick D, eds. Mechanoreceptors specialized to receive tactile information Neuroscience. 2nd ed., Sunderland (MA: ): Sinauer Associates; 2001 [Google Scholar]
- 37.Helios Gene Gun System Instruction Manual , Rev C. Bio-Rad, accessed 2015February9 [Google Scholar]
- 38.Baxter J, Mitragotri S. Jet-induced skin puncture and its impact on needle-free jet injections: experimental studies and a predictive model. J Control Release 2005; 106:361-73; PMID:16002174; http://dx.doi.org/ 10.1016/j.jconrel.2005.05.023 [DOI] [PubMed] [Google Scholar]