

Abstract
We previously reported that MC32 cells resist carcinoembryonic antigen (CEA) DNA vaccination by losing their antigen presentation to Ag-specific CTLs in the context of MHC class I antigens in a colon cancer therapeutic model. In this study, we selected 2 tumor cells, MC32-S2–2 and MC32-S4–2, which have the ability to form tumors in CEA DNA vaccine-immunized mice. Wild type MC32 cells grew significantly less in CEA-immunized mice (with Ag-specific CTL lytic activity) than in control mice (with no Ag-specific CTL lytic activity). However, MC32-S2–2 and MC32-S4–2 cells grew at a similar rate in both control and CEA-immunized mice, confirming their resistant status against CEA DNA vaccination. MC32-S2–2 and MC32-S4–2 cells were not susceptible to lysis by CEA-specific CD8+ T cells. Moreover, when MC32-S2–2 and MC32-S4–2 cells were used as stimulating agents of CEA-specific immune cells for IFN-γ production, these cells failed to stimulate the induction of Ag-specific IFN-γ, suggesting a loss of tumor cell recognition by Ag-specific immune cells. However, MC32-S2–2 and MC32-S4–2 cells expressed MHC class I antigens in a manner similar to that of wild type MC32 cells. Finally, Western blot assay confirmed that in MC32-S2–2 and MC32-S4–2 cells, CEA expression remained absent but mouse CEA was expressed. Taken together, these data show that MC32 cells may also be able to achieve resistance to CEA-specific CTLs by antigen loss in this model.
Keywords: Antitumor immunity, CEA, colon cancer, DNA vaccines, immune evasion
Abbreviations
- CEA
carcinoembryonic antigen
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- EP
electroporation
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HLA
human leukocyte antigen
- IM
intramuscular
- i.v.
intravenously
- LDH
lactate dehydrogenase
- PBS
phosphate-buffered saline
- s.c.
subcutaneously
- PCR
polymerase chain reaction
- UV
ultraviolet
Introduction
Despite the improved therapy modalities for colorectal cancer, a high percentage of patients still develop tumor recurrence after the complete surgical removal of a primary tumor.1 In patients with colorectal cancer, carcinoembryonic antigen (CEA) is over-expressed; CEA is thus being used as a diagnostic marker for post-surgery cancer recurrence and as a predictive marker for cancer progression before surgery.2-4 The self-protein, CEA could be utilized in therapeutic vaccines against colon cancer.5,6 In animal models, human CEA-based vaccines have induced Ag-specific CD8+ T cell responses and antitumor protective activity against Ag-expressing tumor cells.7-9 Furthermore, human CEA DNA vaccines in combination with IL-12 plasmid DNAs augmented Ag-specific antibody and cellular responses.10,11 In that study, both CD8+ T cells and perforin were responsible for tumor cell apoptosis, as demonstrated by immune cell subset depletion and knockout animal studies. Additionally, CEA DNA vaccines are capable of breaking immunological tolerance in a human leukocyte antigen (HLA)-A2.1 transgenic/human CEA hybrid mouse model, thus conferring antitumor protective effects against human CEA-expressing tumor cells.12 Furthermore, in both wild type and CEA transgenic mice, a CEA DNA prime-adenovirus boost vaccination strategy results in the induction of Ag-specific antibody and CD8+ T cell responses and a delay in tumor growth in a CD8+ T cell-dependent manner.13,14 Taken together, these pre-clinical studies suggest that CEA might be useful as a vaccine target to induce Ag-specific CD8+ T cell responses for controlling colon cancer and other cancers expressing CEA.
Tumor cells are heterogeneous and tend to develop numerous mechanisms to escape the host's CTL-mediated immune surveillance.15 Immune-resistant tumor cells appear to be selected after long-term antitumor immunotherapy or even under immune selection conditions. Tumor cells release TGF-β, which inhibits CTL functions, and blocking of TGF-β improves CTL-mediated tumor control.16,17 Complete antigen loss or a lack of antigen epitope presentation on the surface of tumor cells is also responsible for tumor cell insensitivity to tumor Ag-specific CTL-mediated apoptosis.18,19 cFLIP Over-expression has been related to tumor cell resistance to CTL-mediated apoptosis by blocking the death receptor-dependent tumor cell lysis pathway.20 The serine protease inhibitor PI-9r/SPI-6 (an inactivator of granzyme B) also renders tumor cells insensitive to Ag-specific CTL-mediated apoptosis by blocking the granzyme B protein (a primary effector molecule in the perforin/granzyme B-mediated killing).21 Moreover, an increase in the expression of anti-apoptotic molecules is also associated with tumor cell resistance to CTL-mediated tumor cell apoptosis.22 Recently, we also reported that MC32 cells may evade Ag-specific CTL-mediated immune surveillance by altering antigen presentation on the tumor cell surface.9 Taken together, these previous studies including ours show that tumor cells may often become immune-resistant cells that do not respond to Ag-specific CTLs. Therefore, determining the immune escape mechanism(s) is important for designing counter-measures for this CTL resistance.
In this study, we selected MC32-S2–2 and MC32-S4–2 cells with the ability to resist CEA DNA vaccination. We found that these cells were not susceptible to the lysis of CEA-specific CD8+ T cells. Moreover, when MC32-S2–2 and MC32-S4–2 cells were used as stimulating agents of CEA-specific immune cells for IFN-γ production, these cells failed to stimulate Ag-specific IFN-γ production, suggesting a possible defect in tumor cell recognition by Ag-specific immune cells. However, MC32-S2–2 and MC32-S4–2 cells expressed MHC class I antigens in a manner similar to that of wild type MC32 cells. Finally, Western blot assay confirmed that human CEA expression remained absent in the 2 cells. Taken together, these data show that MC32 cells may also achieve resistance to CEA-specific CTLs by losing an antigen in this model.
Results
Selection of MC32-S2–2 and MC32-S4–2 cells with the ability to resist CEA DNA vaccination
Anti-4–1BB Abs were effective at augmenting the Ag-specific CTL lytic and antitumor therapeutic activity of tumor vaccines in 2 of our previous animal studies.23,24 We also tested anti-4–1BB Abs in a CEA DNA vaccine model to determine their antitumor effectiveness. As observed in Figure 1A, animals immunized with pCEA+anti-4–1BB Abs displayed slightly greater antitumor protective responses to MC32 cell challenges compared with those animals immunized with pCEA+control Abs, suggesting that anti-4–1BB Abs might have minimal effects on CEA DNA vaccine-driven tumor control. Specifically, 3 of 8 mice immunized with pCEA plus control IgG formed no tumors after 50 d after tumor cell challenges, and 4 of 8 mice immunized with pCEA plus anti-4–1BB Abs formed no tumors (Fig. 1A). To evaluate the nature of the tumors formed in CEA-immune mice, we surgically removed tumor tissues from 4 mice with tumors that had been immunized with pCEA plus anti-4–1BB Abs. After several rounds of tumor cell culture in vitro, we designated the cells as MC32-S1, MC32-S2, MC32-S3 and MC32-S4. Simultaneously, 4 tumor-free mice (immunized with pCEA plus anti-4–1BB Abs) were re-immunized by intramuscular (IM)-electroporation (EP) with pCEA at 60 d after tumor cell challenges. At 7 d post-final immunization, the 4 mice were re-challenged with wild type MC32 cells on the left flank and with MC32-S1, MC32-S2, MC32-S3 and MC32-S4 cells on the right flank. As shown in Figure 1B–E, wild type MC32 cells and all 4 types of tumor cells formed tumors in age-matched naïve control mice. However, only MC32-S2 and MC32-S4 cells, and not MC32-S1 and MC32-S3 cells, formed tumors in the re-immunized mice (Fig. 1F–I). Next, we surgically removed tumor tissues from the mice with MC32-S2 and MC32-S4 tumors. After several rounds of tumor cell culture in vitro, we designated the cells as MC32-S2–1 and MC32-S4–1. The 3 mice (immunized with pCEA plus control Abs in Fig. 1A) that had been re-immunized twice with pCEA, along with 3 age-matched naïve control mice, were subsequently challenged with these tumor cells. Specifically, each of the 3 mice was challenged with MC32 cells on the upper right flank, MC32-S2–1 cells on the upper left flank and MC32-S4–1 cells on the lower right flank. As shown in Figure 2A–C, MC32, MC32-S2–1 and MC32-S4–1 cells formed tumors in all 3 naïve mice. However, MC32, MC32-S2–1 and MC32-S4–1 cells all failed to form tumors in the 2 re-immunized mice (Fig. 2D and E). However, MC32-S2–1 and MC32-S4–1 cells, but not MC32 cells, formed tumors in the remaining 1 re-immunized mouse (Fig. 2F). Then, we surgically removed tumor tissues from the MC32-S2–1 and MC32-S4–1 tumor-bearing mouse. After several rounds of tumor cell culture in vitro, we designated these cells as MC32-S2–2 and MC32-S4–2.
Figure 1.

Selection of MC32-S2–1 and MC32-S4–1 cells showing tumor growth in mice immunized with CEA DNA vaccines. (A) Each group of mice (n = 6–8/group) was immunized by IM-EP with 50 μg of CEA DNA vaccines (pCEA) per mouse at 0 and 1 weeks. The animals were also injected intraperitoneally with 50 μg of anti-4–1BB Abs at 0 and 1 weeks. At 3 weeks, the mice were challenged s.c. with 1 × 105 MC32 cells per mouse. Tumor sizes were measured and recorded at 41 d following tumor cell challenge. In the group immunized with pcDNA3+anti-4–1BB Abs, one mouse showing no tumor formation was not included in the tumor size calculation to reduce variation. The values and bars represent mean tumor sizes and the SD, respectively. The numbers in (/) denote the number of mice showing no tumor formation at 50 d post-tumor cell challenge/the number of mice tested. Of the 8 mice challenged with MC32 cells following immunization with pCEA+anti-4–1BB Abs, 4 tumor-forming mice were sacrificed. Tumor tissues were surgically removed and then expanded more than 5 times in cDMEM. The resultant 4 tumor cells were designated as MC32-S1, MC32-S2, MC32-S3 and MC32-S4. Of the 8 mice challenged with MC32 cells following immunization with pCEA+anti-4–1BB Abs, 4 tumor-free mice were re-immunized with pCEA at 60 d post-tumor cell challenge. At 7 d following final immunization, each of the mice (F–I) was re-challenged with 5 × 105 MC32 cells per mouse on the right flank and with 5 × 105 MC32-S1 (F), MC32-S2 (G), MC32-S3 (H), or MC32-S4 (I) cells per mouse on the left flank. Each of the control mice was also challenged with 5 × 105 MC32 cells per mouse on the right flank and with 5 × 105 MC32-S1 (B), MC32-S2 (C), MC32-S3 (D) or MC32-S4 (E) cells per mouse on the left flank. Tumor sizes were measured at each time point, and each tumor volume was recorded. *P < 0.05 using ANOVA compared with pcDNA3.
Figure 2.
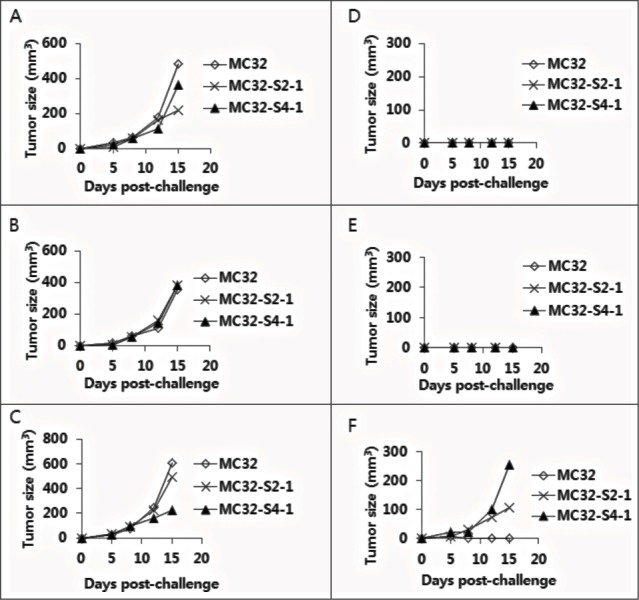
Selection of MC32-S2–2 and MC32-S4–2 cells with resistance to CEA DNA immunization. (A–F) Of the 8 mice challenged with MC32 cells following immunization with pCEA+control Abs (of Fig. 1A), 3 tumor-free mice were re-immunized with pCEA at 60 and 106 d post-tumor cell challenge. At 3 weeks following the final re-immunization, each of the 3 mice (D–F) was re-challenged with 5 × 105 MC32 (on the upper right flank), MC-32-S2–1 (on the lower right flank) or MC-32-S4–1 (on the upper left flank) cells per mouse, in parallel with naïve control mice (A–C). Tumor sizes were measured at each time point, and each tumor volume was recorded.
MC32-S2–2 and MC32-S4–2 cells were resistant to CEA-specific CTL-mediated apoptosis and were insensitive to CEA-specific immune cells for IFN-γ induction
Because we observed that MC32-S2–2 and MC32-S4–2 cells were resistant to CEA DNA immunization, we hypothesized that these tumor cells might be altered such that these cells became resistant to CEA-specific CTL-mediated apoptosis. As shown in Figure 3A, MC32 cells were sensitive to CEA-specific CTL effector cell-mediated apoptosis, whereas MC32-S2–2 and MC32-S4–2 cells were insensitive to CEA-specific CTL effector cell-mediated apoptosis in a manner similar to that of control MC38 cells (lacking CEA expression). We also tested whether these tumor cells might have lost the capacity to activate CEA-specific CD8+ T cells in vitro. When ultraviolet (UV)-inactivated wild type MC32 cells were used as a stimulating agent to CEA-specific immune cells (induced by pCEA immunization), these cells significantly induced IFN-γ production in Ag-specific immune cells (Fig. 3B). However, when UV-inactivated MC32-S2–2 and MC32-S4–2 cells were tested as stimulating agents, these cells failed to induce IFN-γ production in CEA-specific immune cells in a manner similar to that of UV-inactivated MC38 control cells. These in vitro data suggest that MC32-S2–2 and MC32-S4–2 cells might have a defect in presenting an antigen to Ag-specific CTLs, thereby becoming insensitive to antitumor CTL immunity.
Figure 3.
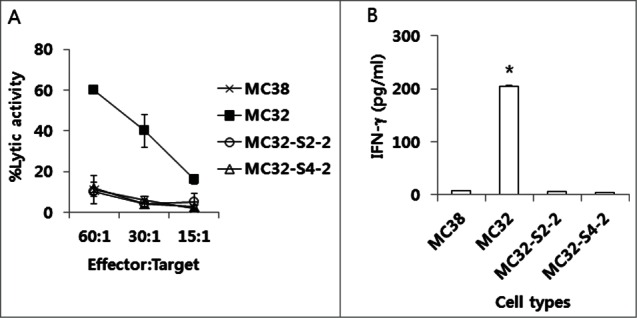
The sensitivity of MC32-S2–2 and MC32-S4–2 cells to CEA-specific CTL-mediated apoptosis in vitro and their ability to stimulate CEA-specific immune cells in vitro. (A) Naïve mice were immunized by IM-EP with 50 μg of CEA DNA vaccines (pCEA) per mouse at 0, 1 and 2 weeks. The mice were sacrificed at 4 weeks, and the spleens were removed for immune cell isolation. The cells were stimulated in vitro with CEA class I peptides and rIL-2 and then reacted with MC32, MC32-S2–2 and MC32-S4–2 cells for in vitro CTL lytic assay, as described in the Materials and Methods. (B) Similar experiments in Fig. 3A, except that 6 × 106 immune cells were stimulated for 2 d with 2 × 106 MC38, MC32, MC32-S2–2 and MC32-S4–2 cells, which had been exposed to UV light for 3 h before immune cell stimulation. Cell supernatants were used for sandwich ELISA to measure IFN-γ. *P < 0.05 using ANOVA compared with MC38.
CEA expression was lacking in MC32-S2–2 and MC32-S4–2 cells
Because we observed that MC32-S2–2 and MC32-S4–2 cells were resistant to CEA-specific CTL-mediated apoptosis and were insensitive to CEA-specific immune cells for IFN-γ induction, we postulated that these tumor cells might not be recognized by Ag-specific CTLs due to a loss of MHC class I antigen expression. To test this possibility, we measured the expression levels of MHC class I antigens. As shown in Figure 4A, MC32-S2–2 and MC32-S4–2 cells expressed MHC class I antigens at levels similar to those of MC32 cells. These data suggest that a loss of MHC class I expression is not associated with this tumor cell resistance. Next, we tested whether the tumor cells might still express human CEA antigens. As shown in Figure 4B, MC32 cells expressed human CEA antigens. However, MC32-S2–2 and MC32-S4–2 cells failed to express human CEA antigens. Instead, all the tested tumor cells expressed murine CEA antigens. These data suggested that MC32-S2–2 and MC32-S4–2 cells lost the human CEA antigen, becoming insensitive to CEA-specific CTL-mediated apoptosis. Next, we tested using polymerase chain reaction (PCR) analysis whether MC32-S2–2 and MC32-S4–2 cells might still possess human CEA genes on the chromosomes. As seen in the Figure 4C, MC32-S2–2 and MC32-S4–2 cells showed the existence of human CEA genes on the chromosomes in a manner similar to MC32 cells. In contrast, MC38 cells displayed no human CEA genes. These data show that a lack of human CEA expression might be resultant from the negative regulation of human CEA expression at the transcriptional and/or post-transcriptional levels.
Figure 4.
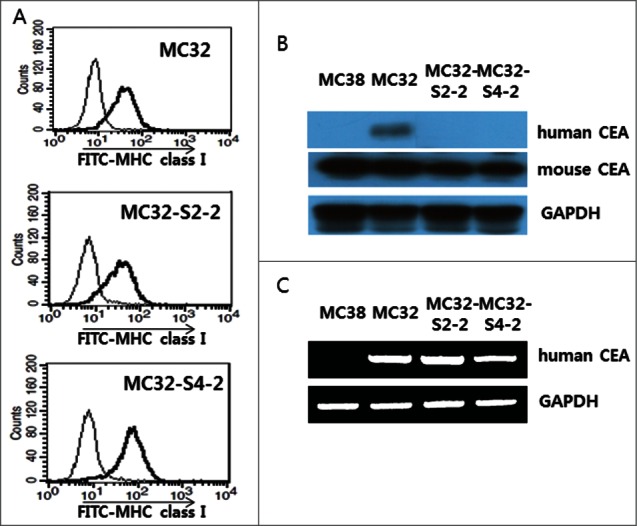
Expression of an MHC class I molecule in MC32-S2–2 and MC32-S4–2 cells and the presence of human CEA in these cells. (A) MC32, MC32-S2–2 and MC32-S4–2 cells were trypsin-treated and then reacted with anti-H-2Kb-FITC. The cells were subsequently analyzed using a flow cytometer. Thin line, cells reacted with FITC-conjugated control Abs; thick line, cells reacted with anti-H-2Kb-FITC. (B) MC38, MC32, MC32-S2–2 and MC32-S4–2 cells were grown in cDMEM. The cells were lysed in RIPA buffer, and 30 μg samples of cell lysates were separated by SDS-PAGE and analyzed by Western blot assay, as described in the Materials and Methods. (C) MC38, MC32, MC32-S2–2 and MC32-S4–2 cells were lysed for genomic DNA purification. Four hundred ng of genomic DNA was tested for PCR assay against human CEA, as described in the Materials and Methods.
MC32-S2–2 and MC32-S4–2 cells were resistant to CEA DNA immunization in vivo
Because we observed that MC32-S2–2 and MC32-S4–2 cells were resistant to CEA DNA vaccination, we next evaluated this resistance in animals. For this evaluation, we immunized mice with CEA DNA vaccines and then challenged these mice with MC32, MC32-S2–2 and MC32-S4–2 cells. Subsequently, we measured tumor sizes and in vivo CTL activity at 13 d post-tumor cell challenge. As shown in Figure 5A, MC32 cells formed significantly smaller tumors in CEA DNA vaccine-immunized mice than in control mice, which is suggestive of the sensitivity of MC32 cells to CEA DNA vaccination. However, MC32-S2–2 and MC32-S4–2 cells formed similar size tumors in both CEA DNA vaccine-immunized and control mice. In terms of in vivo Ag-specific CTL activity, MC32, MC32-S2–2 and MC32-S4–2 tumor-bearing control mice displayed no Ag-specific CTL activity (Fig. 5B, C, D, and H). In contrast, MC32, MC32-S2–2 and MC32-S4–2 tumor-bearing mice previously immunized with CEA DNA vaccines equally exhibited significantly greater CTL lytic activity than the tumor-bearing control mice (Fig. 5E, F, G, and H). These collective data regarding tumor sizes and in vivo CTL activity corroborate the notion that neither MC32-S2–2 nor MC32-S4–2 cells are susceptible to Ag-specific CTL-mediated apoptosis through a loss of CEA expression. This finding was further supported in animal studies. As shown in Figure 6B and D, MC32 cells formed significantly smaller tumors than MC32-S2–2 and MC32-S4–2 cells over time in CEA DNA vaccine-immunized mice. However, MC32, MC32-S2–2 and MC32-S4–2 cells displayed no significant difference in tumor growth over time in control mice (Fig. 6A and C). Taken together, these in vivo data confirm that MC32-S2–2 and MC32-S4–2 cells can evade CEA-specific CTL-mediated apoptosis by antigen loss, which is responsible for a lack of tumor growth inhibition in mice immunized with CEA DNA vaccines.
Figure 5.
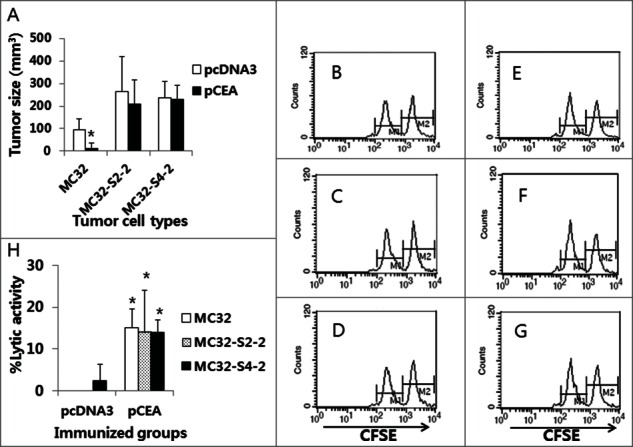
The levels of antitumor protective responses and Ag-specific CTL lytic activity in CEA DNA-immunized mice following a challenge with MC32, MC32-S2–2 and MC32-S4–2 cells. (A) Each group of mice (n=5/group) was immunized by IM-EP with 50 μg of CEA DNA vaccines (pCEA) per mouse at 0 and 1 weeks. At 3 weeks, the animals were challenged s.c. with 1 × 105 MC32, MC32-S2–2 and MC32-S4–2 cells per mouse, along with control mice injected with pcDNA3. Tumor sizes were measured at 13 d post-challenge, and mean tumor volumes were recorded. The values and bars represent mean tumor sizes and the SD, respectively. (B–G) The mice were subsequently tested for the levels of in vivo CTL lytic activity (B, control mice challenged with MC32 cells; C, control mice challenged with MC32-S2–2 cells; D, control mice challenged with MC32-S4–2 cells; E, pCEA-immunized mice challenged with MC32 cells; F, pCEA-immunized mice challenged with MC32-S2–2 cells; G, pCEA-immunized mice challenged with MC32-S4–2 cells) at 13 d post-challenge. For this experiment, CEA peptide-pulsed (CFSE high) and un-pulsed (CFSE low) splenocytes were injected i.v. into the immunized mice, as described in the Materials and Methods. After 20 h, the mice were sacrificed and the splenocytes were analyzed by FACS to measure the levels of CFSE-labeled cells in each subset. M1, un-pulsed CFSE low population; M2, CEA-pulsed CFSE high population. (H) %lytic activity of the tested groups. The values and bars represent mean CTL activity and the SD, respectively. *P < 0.05 using the independent sample's t test compared with MC32 or pcDNA3.
Figure 6.
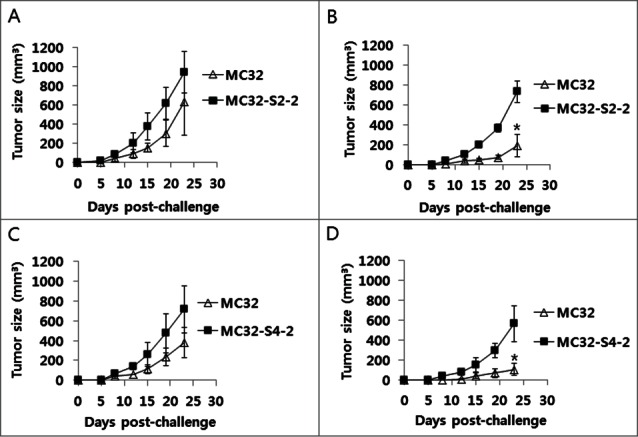
A lack of antitumor protective responses to MC32-S2–2 (A) and MC32-S4–2 cells in CEA DNA-immunized mice. Mice were immunized by IM-EP with CEA DNA vaccines at 0 and 1 weeks. At 3 weeks, the mice (B, D) were challenged s.c. with 1 × 105 MC32 cells per mouse on the right flank and with 1 × 105 MC32-S2–2 (B) and MC32-S4–2 cells (D) per mouse on the left flank. Age-matched control mice (A, C) were also challenged s.c. with 1 × 105 MC32 cells per mouse on the right flank and with 1 × 105 MC32-S2–2 (A) and MC32-S4–2 cells (C) per mouse on the left flank. Tumor sizes were measured at each time point and mean tumor volumes were recorded. The values and bars represent mean tumor volumes and the SD, respectively. *P < 0.05 using the independent sample's t test compared with MC32.
Discussion
In the present study, we observed that MC32-S2–2 and MC32-S4–2 cells were able to resist CEA-specific CTL-mediated immune surveillance by antigen loss. We selected the 2 immune-resistant tumor cells through a series of CEA DNA immunization and MC32 tumor cell challenge studies, which likely forced the cells to completely lose an antigen. We clearly found that the 2 cells expressed MHC class I antigens but remained insensitive to CEA-specific CTL-mediated tumor cell lysis. Furthermore, these cells were unable to stimulate Ag-specific immune cells for IFN-γ induction. These in vitro data suggest that MC32-S2–2 and MC32-S4–2 cells might have defects in antigen processing and presentation processes, thus leading to failed recognition by Ag-specific CTLs. However, in a subsequent analysis, the 2 tumor cells expressed no human CEA antigens, suggesting that antigen loss might be responsible for tumor cell resistance to Ag-specific CTLs in this case. On the other hand, our PCR analysis confirmed that MC32-S2–2 and MC32-S4–2 cells still possess human CEA genes on the chromosomes, suggesting that human CEA expression might be negatively regulated in these 2 cells at the transcriptional and/or post-transcriptional levels. These in vitro data are also consistent with the data from animal studies showing that MC32-S2–2 and MC32-S4–2 cells grew at a similar rate in both control (having no CEA-specific CTL lytic activity) and CEA-immunized mice (having CEA-specific CTL lytic activity), whereas wild type MC32 cells grew significantly less in CEA-immunized mice than in control mice. This finding is consistent with that of a previous report indicating that antigen loss is one mechanism by which tumor cells escape immune surveillance.18 However, our present observation is somewhat distinct from that of our previous report demonstrating that MC32 cells became completely resistant to Ag-specific CTL lytic activity by losing antigen expression on the surface of tumor cells in the context of MHC class I antigens.9 MC32 cells might be genetically unstable and become vulnerable to any possible alterations, including antigen processing and presentation, in the context of MHC class I antigens. However, under severe immune selection conditions, MC32 cells most likely choose a different mechanism for CTL escape, namely, antigen loss. In this case, a loss of antigen expression likely leads to the failure of antigen processing and presentation on the tumor cell surface in the context with MHC class I molecules, resulting in no tumor cell recognition and apoptosis by Ag-specific CTLs. This is supported by our observation that Ag-specific CTLs are still reactive in MC32-S2–2 and MC32-S4–2 tumor-bearing animals, as demonstrated by in vivo CTL assay. However, why MC32 cells had to lose their antigen while remaining able to escape Ag-specific CTL-mediated immune surveillance by altering their antigen expression on the tumor cell surface is currently unclear. Notably, immune selection conditions in a TC-1 tumor model allow tumor cells to develop stem-like phenotypes through activating the transcription factor Nanog, making these tumor cells resistant to tumor-specific CTLs.25 Additionally, in our un-published data, 2 B16 melanoma cell types that displayed resistance to Trp2-specific CTL-mediated immune surveillance were selected through severe immune selection processes. In that study, one B16 cell type was able to escape Ag-specific CTL-mediated apoptosis by altering antigen presentation in the context of MHC class I antigens while the other cell type appeared to have alterations in an intracellular event for CTL escape. In view of these findings, each tumor cell type may develop its own mechanism to resist CTL-mediated immune surveillance. Taken together, our findings support the notion that MC32 cells may choose antigen loss as a CTL escape mechanism under severe immune selection conditions, thus resisting CEA DNA vaccination.
Designing counter-measures for immune-evading tumor cells is critical for improving the efficacy of anticancer vaccines. DNA vaccines targeting 2 different antigens, CEA and HER2, have been reported to induce broader immune responses and confer more effective antitumor effects in an animal tumor model.26 Targeting more than one tumor antigen will most likely be more useful when one antigen is lost during long-term immunotherapy. In this study, we also used human CEA as an antigen in a mouse model. This animal model might be not relevant for evaluating the efficacy of human CEA DNA vaccines due to their antigenicity and different epitope selection in the murine system. An HLA-A2.1 transgenic/human CEA hybrid mouse model is known to be more relevant for testing the potency and immune nature of human CEA vaccines.12 Thus, transgenic animal models might be more valuable for testing the efficacy of anticancer vaccines and immune escape events in tumor cells. However, a prime-boost vaccination strategy in a recent clinical trial using HER2/CEA DNA vaccines and HER2/CEA-expressing adenovirus failed to induce any measurable cellular responses to the 2 tested antigens,27 suggesting that more focused studies remain required for optimal immune induction of tumor-associated antigens, including human CEA, which are generally immune-tolerant.
In summary, we observed that the selected tumor cells, MC32-S2–2 and MC32-S4–2 were not responsive to CEA-specific CTLs. The two cells still expressed MHC class I antigens but failed to stimulate CEA-specific immune cells. Western blot assay further confirmed that the 2 cells lost CEA expression. Furthermore, animal studies demonstrated that MC32-S2–2 and MC32-S4–2 cells form tumors in CEA DNA vaccine-immunized mice (having Ag-specific CTL activity) in a manner similar to that of control mice (having no Ag-specific CTL activity), suggesting their resistance status against tumor-reactive CTLs. Taken together, these in vitro and animal results show that MC32 cells may escape Ag-specific CD8+ CTL-mediated immune surveillance through antigen loss in this model.
Materials and Methods
Mice and Tumors
Female 6 week old C57BL/6 mice were purchased from Daehan Biolink Co., Chungbuk, Korea. They received care under the guidelines of the Institutional Animal Care and Use Committee-approved protocols. Murine colon adenocarcinoma cell lines, MC32 (expressing human CEA) and MC38 (lacking human CEA expression) cells were kindly provided from J. Schlom (NCI, Bethesda, USA) and grown in cDMEM medium (10% FBS, 1% L-glutamine, 1% penicillin/streptomycin). The tumor cells were washed 2 times with phosphate-buffered saline (PBS) and injected into mice.
Reagents and immunization
The plasmid DNA, pcDNA3-CEA was previously described and kindly provided.7 For DNA immunization, animals were injected intramuscularly with 50 μg of pcDNA3-CEA (pCEA) per mouse in a final volume of 50 μl in PBS using a 31-gauge needle (BD, Franklin Lakes, NJ) at 1 week intervals, followed by EP at 0.2A for 4 sec using Cellectra® (equipped with 3 needle probes) of VGX International Inc., Seoul, Korea. Plasmid DNA was produced in bacteria and purified by endotoxin-free Qiagen kits according to the manufacturer's protocol (Qiagen, Valencia, CA). Anti-4–1BB Abs were kindly provided by B. Kwon (University of Ulsan, Korea). Control rat IgG was purchased from Sigma. CEA class I CTL peptides (CGIQNSVSA) were purchased from Peptron (Taejon, Korea).
SDS-PAGE and Western blot assay
MC38, MC32, MC32-S2–2 and MC32-S4–2 cells were lysed with RIPA lysis buffer. Thirty μg of cell lysates were tested for SDS-PAGE as previously described.28 In particular, mice were immunized by IM-EP with 50 μg of pCEA twice at one week intervals and bled at 2 weeks. The collected sera were used as a primary antibody to detect the expression of human CEA for Western blot assay. Anti-murine CEA Abs were purchased from Sino Biological Inc. (Beijing, China). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Abs were purchased from Cell Signaling Technology, Inc.
Genomic DNA purification and PCR analysis
For purification of genomic DNA, tumor cells were lysed by treatment with the reaction buffers of the genomic DNA purification kit (Bioneer, Daejon, Korea) in accordance with the manufacturer's protocol. Four hundred ng of the genomic DNA was reacted for 35 PCR cycles (95°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec). The primers for human CEA were designed as TGTGAACCTGAGGCTCAGAAC and CAACCAGCACTCCAATCATGAT (GenBank accession no. M29540.1; 525 bp of the region of 1681 to 2206 nucleotides). This region contains a nucleotide sequence which encodes the CTL epitopes, CGIQNSVSA tested in this study. The primers for GAPDH were designed as AATGTGTCCGTCGTGGATCT and CCCTGT TGCTGTAGCCGTAT (GenBank accession number GU214026.1; 255 bp of the region of 780 to 1035 nucleotides). The final product was separated by gel electrophoresis on a 1.5% agarose gel.
IFN-γ assay
A 1.5 ml aliquot containing 6 × 106 splenocytes was added to wells of 24 well plates containing 2 × 106 tumor cells, which had been exposed to UV light for 3 h prior to immune cell stimulation. After 2 d incubation, cell supernatants were secured. The obtained cell supernatants were used for detecting levels of IFN-γ using commercial cytokine kits (BD) by adding the extracellular fluids to the IFN-γ-specific ELISA plates.
In vivo and in vitro CTL lytic activity assays
For in vivo CTL lytic assay, spleen cells from naïve mice were treated with red blood cell lysis buffer (Sigma). One fraction of the splenocytes was then pulsed with 5 μg of CEA class I peptides (CGIQNSVSA) in cRPMI for 60 min at 37°C, while the other fraction was left un-pulsed. To generate peptide-pulsed cells with high carboxyfluorescein diacetate succinimidyl ester (CFSE), the peptide-pulsed splenocytes were incubated with 20 μM CFSE in RPMI (2.5% FBS) for 15 min. The un-pulsed cells were instead incubated with 2.5 μM CFSE in RPMI (2.5% FBS) for 15 min to generate non-peptide-pulsed cells with low CFSE. The cells were then washed 3 times with PBS to remove unbound CFSE. Finally, an equal number of pulsed and un-pulsed cells (a total of 2 × 107 cells/0.4 ml/mouse) were injected intravenously (i.v.) into the tested mice. After 20 h, the mice were sacrificed and the spleens were collected. After lysing the red blood cells, the splenocytes were analyzed directly for the 2 cell populations with CFSE staining (CFSE low versus CFSE high) using a flow cytometer (BD). The percentage of lysed cells (%lysis) was calculated as 100 × [1-(runprimed/rprimed)]. The ratio (r) was calculated as %CFSElow/%CFSEhigh. For in vitro CTL lytic assay, splenocytes were cultured with 1 μg/ml of CEA class I peptides (CGIQNSVSA) and 25 units/ml of rIL-2 (BD). The CTL assay was performed in 96-well round bottom plates. Cytolysis was determined by quantitatively measuring lactate dehydrogenase (LDH) activity as previously reported.29-31 Splenocytes served as an effector cell and MC38, MC32, MC32-S2–2 and MC32-S4–2 cells were used as target cells. CTL assay was performed with effector and target cells (1.3 × 104/well) mixed at varying effector cell:target cell ratios in a final volume of 200 μl. After 5 h incubation at 37°C, 50 μl of cell supernatant was collected to test the amount of LDH in the cultured medium according to the manufacturer's protocol (Promega, Madison, WI). After background subtraction, the percent lysis was calculated by 100 × [(experimental release-effector spontaneous release-target spontaneous release)/(target maximum release-target spontaneous release)].
Tumor cell challenge
For antitumor studies, 1–5 × 105 MC32 and MC32-derived tumor cells were injected subcutaneously (s.c.) into the flank of C57BL/6 mice. For tumor re-challenge studies, 5 × 105 MC32 tumor cells per mouse were injected s.c. into the left and right flank of C57BL/6 mice, respectively. Mice were monitored twice per week for tumor growth. Tumor size was measured in mm3 using a caliper, and was recorded as mean tumor volume [longest surface length (a) and width (b), (a × b2)/2]. Mice were euthanized when tumor size reached more than 4000 mm3 in mean tumor volume.
Statistical analysis
Statistical analysis was done by one-way ANOVA with post hoc Dunnett's test and independent sample's t test using the SPSS 17.0 software program. Values of experimental groups were compared with values of control group. The p values <0.05 were considered significant.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A2038430).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank VGX Internation Inc./Inovio providing Cellectra® for this study. We would also like thank H. Kim, K.T. Han and S.N. Danishmalik for their technical assistance for this study.
References
- 1.Manfredi S, Bouvier AM, Lepage C, Hatem C, Dancourt V, Faivre J. Incidence and patterns of recurrence after resection for cure of colonic cancer in a well defined population. Br J Surg 2006; 93:1115-22; PMID:16804870; http://dx.doi.org/ 10.1002/bjs.5349 [DOI] [PubMed] [Google Scholar]
- 2.Thompson J, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal 1991; 5:344-66; PMID:1941355; http://dx.doi.org/ 10.1002/jcla.1860050510 [DOI] [PubMed] [Google Scholar]
- 3.Hara M, Sato M, Takahashi H, Takayama S, Takeyama H. Accuracy of monitoring serum carcinoembryonic antigen levels in postoperative stage III colorectal cancer patients is limited to only the first postoperative year. Surg Today 2011; 41:1357-60; PMID:21922357; http://dx.doi.org/ 10.1007/s00595-010-4519-2 [DOI] [PubMed] [Google Scholar]
- 4.Wang WS, Lin JK, Chiou TJ, Liu JH, Fan FS, Yen CC, Lin TC, Jiang JK, Yang SH, Wang HS, Chen PM. Preoperative carcinoembryonic antigen level as an independent prognostic factor in colorectal cancer: Taiwan experience. Jpn J Clin Oncol 2000; 30:12-6; PMID:10770562; http://dx.doi.org/ 10.1093/jjco/hyd003 [DOI] [PubMed] [Google Scholar]
- 5.Kantor J, Irvine K, Abrams S, Kaufman H, DiPietro J, Schlom J. Antitumor activity and immune responses induced by a recombinant carcinoembryonic antigen-vaccinia virus vaccine. J Natl Cancer Inst 1992; 84:1084-91; PMID:1619682; http://dx.doi.org/ 10.1093/jnci/84.14.1084 [DOI] [PubMed] [Google Scholar]
- 6.Hodge J. Carcinoembryonic antigen as a target for cancer vaccines. Cancer Immunol Immunother 1996; 43:127-34; PMID:9001565; http://dx.doi.org/ 10.1007/s002620050313 [DOI] [PubMed] [Google Scholar]
- 7.Kim SG, Park MY, Kim CH, Sohn HJ, Kim HS, Park JS, Kim HJ, Oh ST, Kim TG. Modification of CEA with both CRT and TAT PTD induces potent anti-tumor immune responses in RNA-pulsed DC vaccination. Vaccine 2008; 26:6433-40; PMID:18812201; http://dx.doi.org/ 10.1016/j.vaccine.2008.08.072 [DOI] [PubMed] [Google Scholar]
- 8.Park JS, Kim HS, Park HM, Kim CH, Kim TG. Efficient induction of anti-tumor immunity by a TAT-CEA fusion protein vaccine with poly(I:C) in a murine colorectal tumor model. Vaccine 2011; 29:8642-8; PMID:21945963; http://dx.doi.org/ 10.1016/j.vaccine.2011.09.052 [DOI] [PubMed] [Google Scholar]
- 9.Ahn E, Kim H, Han KT, Sin JI. A loss of antitumor therapeutic activity of CEA DNA vaccines is associated with the lack of tumor cells' antigen presentation to Ag-specific CTLs in a colon cancer model. Cancer Lett 2015; 356:676-85; PMID:25449428; http://dx.doi.org/ 10.1016/j.canlet.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 10.Song K, Chang Y, Prud'homme GJ. IL-12 plasmid-enhanced DNA vaccination against carcinoembryonic antigen (CEA) studied in immune-gene knockout mice. Gene Ther 2000; 7:1527-35; PMID:11021590; http://dx.doi.org/ 10.1038/sj.gt.3301274 [DOI] [PubMed] [Google Scholar]
- 11.Song K, Chang Y, Prud'homme GJ. Regulation of T-helper-1 vs. T-helper-2 activity and enhancement of tumor immunity by combined DNA-based vaccination and nonviral cytokine gene transfer. Gene Ther 2000; 7:481-92; PMID:10757021; http://dx.doi.org/ 10.1038/sj.gt.3301123 [DOI] [PubMed] [Google Scholar]
- 12.Conforti A, Peruzzi D, Giannetti P, Biondo A, Ciliberto G, La Monica N, Aurisicchio L. A novel mouse model for evaluation and prediction of HLA-A2-restricted CEA cancer vaccine responses. J Immunother 2009; 32:744-54; PMID:19561534; http://dx.doi.org/ 10.1097/CJI.0b013e3181aee1b6 [DOI] [PubMed] [Google Scholar]
- 13.Facciabene A, Aurisicchio L, Elia L, Palombo F, Mennuni C, Ciliberto G, La Monica N. Vectors encoding carcinoembryonic antigen fused to the B subunit of heat-labile enterotoxin elicit antigen-specific immune responses and antitumor effects. Vaccine 2007; 26:47-58; PMID:18055074; http://dx.doi.org/ 10.1016/j.vaccine.2007.10.060 [DOI] [PubMed] [Google Scholar]
- 14.Mennuni C, Calvaruso F, Facciabene A, Aurisicchio L, Storto M, Scarselli E, Ciliberto G, La Monica N. Efficient induction of T-cell responses to carcinoembryonic antigen by a heterologous prime-boost regimen using DNA and adenovirus vectors carrying a codon usage optimized cDNA. Int J Cancer 2005; 117:444-55; PMID:15906358; http://dx.doi.org/ 10.1002/ijc.21188 [DOI] [PubMed] [Google Scholar]
- 15.Igney FH, Krammer PH. Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol 2002; 71:907-20; PMID:12050175 [PubMed] [Google Scholar]
- 16.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-b signaling in T cells. Nat Med 2001; 7:1118-22; PMID:11590434; http://dx.doi.org/ 10.1038/nm1001-1118 [DOI] [PubMed] [Google Scholar]
- 17.Thomas DA, Massagué J. TGF-b directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer cell 2005; 8:369-80; PMID:16286245; http://dx.doi.org/ 10.1016/j.ccr.2005.10.012 [DOI] [PubMed] [Google Scholar]
- 18.Urban JL, Kripke ML, Schreiber HJ. Stepwise immunological selection of antigenic variants during tumor growth. J Immunol 1986; 137:3036-41; PMID:3489784 [PubMed] [Google Scholar]
- 19.Bai XF, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest 2003; 111:1487-96; PMID:12750398; http://dx.doi.org/ 10.1172/JCI17656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med 1999; 190:1033-8; PMID:10510093; http://dx.doi.org/ 10.1084/jem.190.7.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medema JP, de Jong J, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, Franken KL, Hahne M, Albar JP, Melief CJ, Offringa R. Blockade of the granzyme B/ perforin pathway through overexpression of the serine protease inhibitor PtdIns-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc Natl Acad Sci USA 2001; 98:11515-20; PMID:11562487; http://dx.doi.org/ 10.1073/pnas.201398198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jazirehi AR, Baritaki S, Koya RC, Bonavida B, Economou JS. Molecular mechanism of MART-1+/A*0201+ human melanoma resistance to specific CTL-killing despite functional tumor-CTL interaction. Cancer Res 2011; 71:1406-17; PMID:21159666; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1296 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Kim H, Kwon B, Sin JI. Combined stimulation of IL-2 and 4-1BB receptors augments the antitumor activity of E7 DNA vaccines by increasing Ag-specific CTL responses. PLoS One 2013; 8:e83765; PMID:24391824; http://dx.doi.org/ 10.1371/journal.pone.0083765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sin JI, Kim H, Ahn E, Jeon YH, Park WS, Lee SY, Kwon B. Combined stimulation of TLR9 and 4.1BB augments Trp2 peptide vaccine-mediated melanoma rejection by increasing Ag-specific CTLs and their infiltration into tumor sites. Cancer Lett 2013; 330:190-9; PMID:23219755; http://dx.doi.org/ 10.1016/j.canlet.2012.11.045 [DOI] [PubMed] [Google Scholar]
- 25.Noh KH, Lee YH, Jeon JH, Kang TH, Mao CP, Wu TC, Kim TW. Cancer vaccination drives Nanog-dependent evolution of tumor cells toward an immune-resistant and stem-like phenotype. Cancer Res 2012; 72:1717-27; PMID:22337995; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aurisicchio L, Peruzzi D, Koo G, Wei WZ, La Monica N, Ciliberto G. Immunogenicity and therapeutic efficacy of a dual-component genetic cancer vaccine cotargeting carcinoembryonic antigen and HER2/neu in preclinical models. Hum Gene Ther 2014; 25:121-31; PMID:24195644; http://dx.doi.org/ 10.1089/hum.2013.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz-Montero CM, Chiappori A, Aurisicchio L, Bagchi A, Clark J, Dubey S, Fridman A, Fabregas JC, Marshall J, Scarselli E, La Monica N, Ciliberto G, Montero AJ. Phase 1 studies of the safety and immunogenicity of electroporated HER2/CEA DNA vaccine followed by adenoviral boost immunization in patients with solid tumors. J Transl Med 2013; 11:62; PMID:23497415; http://dx.doi.org/ 10.1186/1479-5876-11-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sin JI, Sung JH, Suh YS, Lee AH, Chung JH, Sung YC. Protective immunity against heterologous challenge with encephalomyocarditis virus by VP1 DNA vaccination: effect of co-injection with a granulocyte,macrophage-colony stimulating factor gene. Vaccine 1997; 15:1827-33; PMID:9413089; http://dx.doi.org/ 10.1016/S0264-410X(97)88856-1 [DOI] [PubMed] [Google Scholar]
- 29.Bae SH, Park YJ, Park JB, Choi YS, Kim MS, Sin JI. Therapeutic synergy of human papillomavirus E7 subunit vaccines plus cisplatin in an animal tumor model: causal involvement of increased sensitivity of cisplatin-treated tumors to CTL-mediated killing in therapeutic synergy. Clin Cancer Res 2007; 13:341-9; PMID:17200373; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1838 [DOI] [PubMed] [Google Scholar]
- 30.Ye GW, Park JB, Park YJ, Choi YS, Sin JI. Increased sensitivity of radiated murine cervical cancer tumors to E7 subunit vaccine-driven CTL-mediated killing induces synergistic antitumor activity. Mol Ther 2007; 15:1564-70; PMID:17505485; http://dx.doi.org/ 10.1038/sj.mt.6300149 [DOI] [PubMed] [Google Scholar]
- 31.Sin JI, Park JB, Lee IH, Park D, Choi YS, Choe J, Celis E. Intratumoral electroporation of IL-12 cDNA eradicates established melanomas by Trp2180-188-specific CD8+ CTLs in a perforin/granzyme-mediated and IFN-g dependent manner: application of Trp2180-188 peptides. Cancer Immunol Immunother 2012; 61:1671-82; PMID:22382361; http://dx.doi.org/ 10.1007/s00262-012-1214-8 [DOI] [PMC free article] [PubMed] [Google Scholar]