

Abstract
Background
Ipilimumab is an approved immunotherapy that has shown an overall survival benefit in patients with cutaneous metastatic melanoma in two phase III trials. As results of registrational trials might not answer all questions regarding safety and efficacy of ipilimumab in patients with advanced melanoma seen in daily clinical practice, the Dermatologic Cooperative Oncology Group conducted a phase II study to assess the efficacy and safety of ipilimumab in patients with different subtypes of metastatic melanoma.
Patients and methods
We undertook a multicenter phase II study in melanoma patients irrespective of location of the primary melanoma. Here we present data on patients with pretreated metastatic cutaneous, mucosal and occult melanoma who received up to four cycles of ipilimumab administered at a dose of 3 mg/kg in 3 week intervals. Tumor assessments were conducted at baseline, weeks 12, 24, 36 and 48 according to RECIST 1.1 criteria. Adverse events (AEs), including immune-related AEs were graded according to National Cancer Institute Common Toxicity Criteria (CTC) v.4.0. Primary endpoint was the OS rate at 12 months.
Results
103 pretreated patients received at least one dose of ipilimumab, including 83 cutaneous, seven mucosal and 13 occult melanomas. 1-year OS rates for cutaneous, mucosal and occult melanoma were 38 %, 14 % and 27 %, respectively. Median OS was 6.8 months (95 % CI 5.3–9.9) for cutaneous, 9.6 months (95 % CI 1.6–11.1) for mucosal, and 9.9 months (lower 95 % CI 2.3, upper 95 % CI non-existent) for occult melanoma. Overall response rates for cutaneous, mucosal and occult melanoma were 16 %, 17 % and 11 %, respectively. Eleven patients had partial response (16 %) and ten patients experienced stable disease (14 %), none achieved a complete response. Treatment-related AEs were observed in 71 patients (69 %), including 20 grade 3–4 events (19 %). No new and unexpected safety findings were noted.
Conclusions
Ipilimumab is a treatment option for pretreated patients with advanced cutaneous melanoma seen in daily routine. Toxicity was manageable when treated as per protocol-specific guidelines.
Trial registration: Clinical Trials.gov NCT01355120
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-015-0716-5) contains supplementary material, which is available to authorized users.
Keywords: Phase II, Melanoma, Cutaneous melanoma, Mucosal melanoma, Occult melanoma, Ipilimumab, Anti-CTLA-4
Background
From a clinical like from a scientific perspective, the recent advances in cancer immunotherapy have been acknowledged as a major breakthrough [1]. Especially for melanoma patients, immune checkpoint inhibitors begin to witness an enormous therapeutic potential, resulting very recently in the approval of the first-in-class anti-programmed-death-receptor-1 (PD-1) inhibitors nivolumab and pembrolizumab for the treatment of unresectable or metastatic melanoma [2–5].
The cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) inhibitor ipilimumab has been authorized for use in advanced, metastatic melanoma in the United States and in the European Union—as in many other countries worldwide since—on basis of two pivotal phase III studies [6, 7]. CTLA-4, a native regulator of T cell activation, downregulates T-cell function through a variety of mechanisms, and finally induces T-cell cycle arrest [8]. Because many of the immune checkpoints are regulated by ligand-receptor interactions, CTLA-4 can be easily blocked by monoclonal antibodies or recombinant ligand-like proteins that block CTLA-4 as a negative regulator of immunity, hence enhancing natural antitumor immunity [9].
Serving as the first mechanistically defined immune checkpoint inhibitor, ipilimumab has been intensely investigated in clinical registrational trial settings [6, 7] in patients with cutaneous melanoma, the most frequent melanoma subgroup with morphological and molecular distinctions from other clinical disease subgroups [10]. However, results of registrational trials might not answer all questions regarding safety and efficacy of ipilimumab in advanced melanoma patient cohorts seen in daily routine.
Here we report the results of the open-label, multicenter, single-arm phase II DeCOG trial to further evaluate the efficacy and safety of 3 mg/kg ipilimumab in pretreated patients with cutaneous, mucosal and occult metastatic melanoma seen in daily routine in interdisciplinary skin cancer units in Germany. Data for patients with ocular melanoma are reported elsewhere [11].
Patients and methods
Patients
Eligibility criteria included documented unresectable stage III or stage IV metastatic cutaneous, occult, mucosal and ocular melanoma according to American Joint Committee on Cancer cutaneous melanoma staging criteria [12]. Patients who had received at least one prior systemic therapy were eligible. Previous systemic treatment had to be completed ≥28 days before receiving ipilimumab. Additional requirements included age ≥18 years, Eastern Cooperative Oncology Group (ECOG) performance status ≤2, life expectancy of ≥6 months (estimation of life expectancy was at the discretion of the participating investigators), measurable disease according to Response Evaluation Criteria In Solid Tumors (RECIST) 1.1 [13], adequate bone marrow, renal and hepatic function. Patients with a history of active autoimmune disease and chronic use of systemic corticosteroids were excluded. Patients with asymptomatic, radiographically stable previously treated or untreated brain metastases were eligible.
Study design
This multicenter, open-label, phase II study (DeCOG-MM-PAL11-Trial; CA184-137) was conducted in two parts. Part 1 of the study was open for recruitment from May 2011 to August 2011; in an Additional file 1: Figure S1 the patient flow is described. This part allowed recruitment of pretreated melanoma patients irrespective of location of the primary melanoma. Part 2, which was only eligible for patients with pretreated or treatment-naïve metastatic ocular melanoma to allow for a valid analysis of this subgroup, was closed on September 30, 2012. Data from part 1 and 2 for patients with ocular melanoma are reported elsewhere [11]. Twenty-five Dermatologic Cooperative Oncology Group (DeCOG) skin cancer units in Germany participated. The study was approved by institutional ethics committee University Duisburg-Essen (approval number 10–4531) and the German competent authority Paul-Ehrlich-Institute (Langen, Germany, approval number 1233), and conducted in accordance with the Declaration of Helsinki/Good Clinical Practice. All patients gave written informed consent. The protocol for this trial is available as Additional file 2. Ipilimumab was administered intravenously over 90 min at a dose of 3 mg/kg every 3 weeks for a total of four infusions. Patients with progressive disease (PD) at ≥3 months from week 12 assessment following stable disease (SD), an initial partial (PR) or complete response (CR) were eligible for re-induction with ipilimumab following at the same dosage. Dose reduction was not allowed, but skipping of one dose of ipilimumab was recommended when adverse events (AE) occurred. Rapid disease progression, intolerable toxicity or patient withdrawal led to treatment discontinuation. The primary endpoint was the overall survival (OS) rate at 12 months.
Assessments
Regular assessments, including a physical examination and standardized blood testing, were carried out at baseline and every 3 weeks during induction and re-induction phases. Tumor assessments were conducted at baseline, weeks 12, 24, 36 and 48 using the RECIST version 1.1 [13]. Adverse events (AEs) were graded according to the National Cancer Institute Common Toxicity Criteria (CTC version 4.0). All AEs were recorded from the time of the first ipilimumab administration until 70 days after treatment discontinuation. AEs were defined as an immune-related AE (irAE) if they were associated with drug exposure, consistent with an immune phenomenon and if other causes were ruled out. IrAE management was based on protocol-specific treatment algorithms. All AEs that were definitely, probably or possibly related to study drug were defined as related AEs.
Statistical methods
This report includes results based on the data cutoff of December 6, 2013. Patient and disease characteristics were analyzed using descriptive statistics. Categorical values were expressed as counts and percentage whereas continuous values were expressed as median and range values. OS was defined as the time from the first administration of ipilimumab to death from any cause. Patients last known to be alive were censored at the date of last contact. Progression-free survival (PFS) was defined as the time from the first dose of ipilimumab to the first date of documented progression as per RECIST, or date of death, whichever came first. Patients last known to be alive and progression-free were censored at the date of last contact. PFS rate at 6 months was defined as the proportion of patients being alive and without progress 6 months after the first ipilimumab administration. Patients with unknown survival status or unknown status of progression at 6 months were censored. The 1- and 2-year survival rates were defined as the proportion of patients being alive 12 or 24 months after their first ipilimumab administration. Patients with unknown survival status at 12 or 24 months were censored. OS, PFS, PFS rate at 6 months, 1- and 2-year survival rates were estimated by the Kaplan–Meier method. For medians of OS and PFS, 95 % confidence intervals (CIs) were calculated using the Brookmeyer and Crowley method. The log-rank test was used to compare the 1-year and 2-year OS rates in patients with cutaneous melanoma between several subgroups, i.e. the BRAF mutational status, the presence of brain metastases, the lactate dehydrogenase (LDH) level prior to receiving ipilimumab [<2-fold upper level norm (ULN) vs. ≥2× ULN], the number of ipilimumab doses (<4 vs. 4), and the absolute lymphocyte count (ALC) (<1000/µl vs. ≥1000/µl) before the first (week 1), the second (week 4) and the third dose (week 7) of ipilimumab. Due to small sample sizes comparisons of 1-year and 2-year OS rates in patients with mucosal and occult melanoma were not done. Two sided p values were evaluated and a p value of <0.05 was considered statistically significant. All variables with significant differences between their stratifications regarding the overall survival were included in a multivariate Cox proportional hazards model. To determine potential predictors, all independent covariates (LDH, number of ipilimumab doses, ALC week 4, brain metastases), were entered into a backward Cox regression model for the overall survival. The stay level was p = 0.05. All covariates being still significant were considered as potential predictors. For the hazard ratio, 95 % CIs were calculated using the Wald method. The overall response rate (ORR) was defined as the proportion of patients with PR and CR whereas the disease control rate (DCR) was defined as the proportion of patients with CR, PR and SD. Lost to follow-up was documented if the patient did not respond to phone calls (3 times) and to a written invitation. Analyses were carried out using SAS software, version 9.3 (Cary, NC, USA).
Results
Patients
Between May to August 2011, 103 patients were enrolled and received at least one dose of ipilimumab, including 83 patients with cutaneous melanoma, 13 with occult melanoma and seven with mucosal melanoma (Table 1). Baseline patient characteristics are reported in Table 1. All 103 patients had received previous systemic anti-cancer treatment, including chemotherapy, immunotherapy, and targeted agents (Table 1). The most common chemotherapies were dacarbazine and carboplatin/paclitaxel given in 73 (71 %) and 30 (29 %) of all patients. None had previously received ipilimumab but 18 patients had undergone previous immunotherapy treatment with interferon α or vaccination. 31 patients presented with brain metastases at study entry, with similar proportions observed across the different melanoma subtypes (Table 1). Sixty-four patients (62 %) completed the induction phase, including 52 patients with cutaneous, four with mucosal and eight with occult melanoma. Three patients with cutaneous melanoma experienced PR at week 12 and were re-induced after 91, 232 and 217 days, respectively (Table 2). The median number of doses received in the induction phase was four (range 1–4). Among the 39 patients (38 %) who did not complete the induction phase, 11 (11 %) died, 16 (16 %) developed PD, eight (8 %) had intolerable AEs and four (4 %) withdrew their informed consent.
Table 1.
Baseline patient characteristics (n = 103 patients totally)
| Patient characteristics | Cutaneous melanoma | Mucosal melanoma | Melanoma of unknown primary | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| No. patients, % | 83 | 100 | 7 | 100 | 13 | 100 |
| Age, years | ||||||
| Median (range) | 63 | (29–85) | 63 | (33–37) | 62 | (40–77) |
| Sex | ||||||
| Male | 53 | 64 | 2 | 29 | 11 | 29 |
| Female | 30 | 36 | 5 | 71 | 2 | 71 |
| ECOG baseline | ||||||
| 0 | 51 | 61 | 2 | 29 | 12 | 92 |
| 1 | 23 | 28 | 5 | 71 | 1 | 8 |
| 2 | 9 | 11 | – | – | ||
| BRAF mutation | ||||||
| Not mutated | 29 | 35 | 3 | 43 | 5 | 39 |
| Mutated | 17 | 21 | – | 6 | 46 | |
| Not known | 37 | 45 | 4 | 57 | 2 | 15 |
| Disease stage (all: Stage IV) | ||||||
| M1a | 6 | 7 | – | 3 | 23 | |
| M1b | 15 | 18 | 2 | 29 | 1 | 8 |
| M1c | 62 | 75 | 5 | 71 | 9 | 69 |
| LDH | ||||||
| <2 ULN | 67 | 81 | 5 | 71 | 11 | 85 |
| ≥2 ULN | 16 | 19 | 2 | 29 | 2 | 15 |
| Brain metastases | ||||||
| No | 57 | 69 | 5 | 71 | 10 | 77 |
| Yes | 26 | 31 | 2 | 29 | 3 | 23 |
| Prior systemic therapy in stage IV (except radiotherapy) | ||||||
| No | – | – | – | |||
| Yes | 83 | 100 | 7 | 100 | 13 | 100 |
| Number of prior systemic therapies | ||||||
| 1 | 42 | 51 | 6 | 86 | 8 | 62 |
| 2 | 27 | 33 | – | 3 | 23 | |
| ≥3 | 13 | 16 | 1 | 14 | 2 | 15 |
| Not applicable | 1 | 1 | – | – | ||
| Immunotherapy | ||||||
| No | 67 | 81 | 7 | 100 | 11 | 85 |
| Yes | 16 | 19 | – | 2 | 15 | |
| If yes, type of previous immunotherapy | ||||||
| Interferon alpha | 11 | 13 | – | 2 | 15 | |
| Vaccination | 5 | 6 | – | – | ||
| Kinase inhibitors | ||||||
| No | 71 | 86 | 7 | 100 | 9 | 69 |
| Yes | 12 | 14 | – | 4 | 31 | |
| If yes, type of previous kinase inhibitor | ||||||
| BRAF inhibitor | 7 | 8 | – | 2 | 15.5 | |
| MEK inhibitor | 4 | 5 | 2 | 15.5 | ||
| Chemotherapy | ||||||
| 0 | 9 | 11 | – | 2 | 15 | |
| 1 | 47 | 57 | 6 | 86 | 8 | 62 |
| 2 | 20 | 24 | – | 2 | 15 | |
| ≥3 | 7 | 8 | 1 | 14 | 1 | 8 |
ECOG Eastern Cooperative Oncology Group, LDH lactate dehydrogenase
Table 2.
Outcomes of patients with ipilimumab re-induction therapy
| Age, years | Best response at week 12 (RECIST) | Duration between 1st restaging (week 12) and re-induction therapy (days) | Response at 1st restaging after re-induction (RECIST) | Best overall response after re-induction (RECIST) | Time from 1st dose to death/follow-up (months) | Alive |
|---|---|---|---|---|---|---|
| 74 | PR | 91 | SD | SD | 17.1 | Yes |
| 56 | PR | 232 | PR | CR | 25.5 | Yes |
| 73 | PR | 217 | PD | SD | 24.8 | Yes |
RECIST response evaluation criteria in solid tumors, PR partial response, SD stable disease, PD progressive disease
Efficacy
The 1-year rate for OS was 38 % (95 % CI 27–49) for cutaneous melanoma, 14 % (95 % CI 1–47) for mucosal melanoma, and 27 % (95 % CI 5–57) for occult melanoma. 2-year OS rates for cutaneous and occult melanoma were 22 % (95 % CI 13–33) and 27 % (95 % CI 5–57), respectively. All of the patients with mucosal melanoma died before month 24 after the first ipilimumab administration. Six-month rate for PFS were 16 % (95 % CI 9–25) for cutaneous melanoma, 14 % (95 % CI 1–47) for mucosal melanoma, and 17 % (95 % CI 3–41) for occult melanoma. Median OS from the first dose of ipilimumab for cutaneous, mucosal and occult melanoma were 6.8 (95 % CI 5.3–9.9; Fig. 1a), 9.6 (95 % CI 1.6–11.1; Fig. 1b) and 9.9 (lower 95 % CI 2.3, upper 95 % CI non-existent; Fig. 1c) months, respectively. Seventy of 103 patients were evaluable for efficacy assessment (Table 3). Among the 33 patients (32 %) who were not assessable, 22 died before the assessment of change in tumor burden (including 13 with brain metastases), three developed PD (including two with brain metastases), three had intolerable AEs, one had no measurable disease at baseline, three withdrew their informed consent (including one with brain metastases) and one was lost to follow-up. The DCR was 29 % for cutaneous, 50 % for mucosal and 22 % for occult melanoma (Table 3). Overall response rates for cutaneous, mucosal and occult melanoma were 16 %, 17 % and 11 %, respectively (Table 3). Among the 70 patients evaluable for response, the overall response rate for 15 patients with brain metastases was 13 %: a response rate similar to the one found for the remaining 55 patients without brain metastases (16 %). Of the 15 patients with brain metastases seven patients had intracranial SD, seven intracranial PD and one patient experienced intracranial CR. In total, ten patients showed similar response pattern in intracranial and extracranial metastases and five patients had different response pattern, e.g. in one patient an intracranial response (CR) was observed, unfortunately associated with extracranial PD (Additional file 3: Table S3).
Fig. 1.

Kaplan–Meier curves for 1-year overall survival (OS) rates of different melanoma subtypes. Pretreated patients with a metastatic cutaneous melanoma (1-year OS rate: 38 %), b mucosal melanoma (1-year OS rate: 14 %), and c occult melanoma (1-year OS rate: 27 %). All patients received ipilimumab 3 mg/kg
Table 3.
Overall response rates (ORR) and disease control rates (DCR) (n = 70 patients totally)
| Patients with measurable disease (and at least one tumor assessment) | Cutaneous melanoma | Mucosal melanoma | Melanoma of unknown primary | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| No. patients (%) | 55 | 100 | 6 | 100 | 9 | 100 |
| Response pattern (acc. to RECIST) | ||||||
| Complete response | – | – | – | |||
| Partial response | 9 | 16 | 1 | 17 | 1 | 11 |
| Stable disease | 7 | 13 | 2 | 33 | 1 | 11 |
| Progressive disease | 39 | 71 | 3 | 50 | 7 | 78 |
| Best ORR (according to RECIST) | ||||||
| ORR (=CR + PR) | 9 | 16 | 1 | 17 | 1 | 11 |
| ORR at week 12 | 7 | 13 | 1 | 17 | – | |
| ORR at week 24 | 6 | 11 | 1 | 17 | – | |
| Best DCR (according to RECIST) | ||||||
| DCR (=CR + PR + SD) | 16 | 29 | 3 | 50 | 2 | 22 |
| DCR at week 12 | 15 | 27 | 3 | 50 | 2 | 22 |
| DCR at week 24 | 10 | 18 | 1 | 17 | 1 | 11 |
CR complete response, PR partial response, RECIST response evaluation criteria in solid tumors, SD stable disease
The 1-year OS rate was higher in patients with cutaneous melanoma who had no brain metastases (51 % vs. 12 %, p < 0.0001, Fig. 2a), in patients with a LDH level <2× ULN (42 % vs. 19 %, p = 0.0007), in patients who received four ipilimumab doses (53 % vs. 14 %, p < 0.0001; Fig. 2b), and in patients with an ALC ≥1000/µl before the second dose of ipilimumab (week 4) (47 % vs. 22 %, p = 0.002; Fig. 2c). The apparent better OS observed in patients who received all four ipilimumab doses, could be solely based on a time dependent bias, as receiving four doses of ipilimumab required surviving >10 weeks after therapy initiation. This only applied to 39 % of the patients who received <4, but not surprisingly all with four doses of ipilimumab. BRAF mutational status, the ALC before the first and the third dose of ipilimumab in patients with cutaneous melanoma were not associated with OS. In a multivariate analysis, the factors independently associated with better OS were the administration of four ipilimumab doses (e.g. patients with less than 4 doses were at higher risk of death; hazard ratio 4.3, 95 % CI 2.3–8.0), an ALC ≥1000/µl before the second dose of ipilimumab (week 4) (e.g. patients with ALC <1000/µl were at higher risk of death; hazard ratio 2.0; 95 % CI 1.1–3.8), and the absence of brain metastases (e.g. patients with brain metastases were at higher risk of death; hazard ratio 1.9, 95 % CI 1.0–3.5).
Fig. 2.
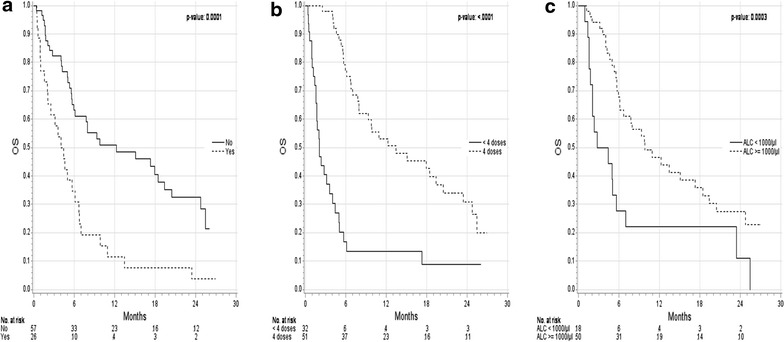
Kaplan–Meier curves for overall survival (OS) of subgroups (pretreated patients with metastatic cutaneous melanoma). Subgroups were stratified as follows: by a the absence of brain metastases before the first dose of ipilimumab; Absence of brain metastases: median OS 12.3 months (95 % CI 6.0–19.4); brain metastases present: median OS 4.2 months (95 % CI 2.0–6.1); b the number of ipilimumab doses (4 versus <4); 4 doses: median OS 13.5 months (95 % CI 7.9–20.4); <4 doses: median OS 2.1 months (95 % CI 1.6–4.1); and c the absolute lymphocyte count (ALC) (≥1000/µl versus <1000/µl) before the second dose (i.e. week 4) of ipilimumab; ALC ≥1000/µl: median OS 9.9 months (95 % CI 6.1–18.5); ALC <1000/µl: median OS 3.6 months (95 % CI 1.8–5.6)
Safety
Ninety-eight of 103 patients (95 %) experienced one or more AEs (Table 4). Treatment-related AEs were reported in 71 patients (69 %); 20 patients (19 %) had treatment-related grade 3 or 4 AEs. The majority of treatment-related AEs were irAEs, occurring in 52 patients (51 %). Most common irAEs were gastrointestinal disorders—diarrhea and colitis, skin-related toxic effects—pruritus and rash, and hepatic disorders—increased alanine aminotransferases (ALT) and aspartate aminotransferases (Table 4). The most frequent grade 3 or 4 irAEs were diarrhea and colitis, noted in ten (10 %) and 5 patients (5 %), respectively. There was one patient with a gastrointestinal perforation due to grade 3 colitis and diarrhea. After surgery and treatment with 2 mg/kg methylprednisolone intravenous, diarrhea and colitis improved and corticosteroid therapy was tapered slowly over 6 weeks. Immune-related AEs were generally reversible when managed as per protocol-specific treatment guidelines. Most of the irAEs resolved with corticosteroid therapy. None of the patients required additional immunosuppression with infliximab or mycophenylate mofetil. Treatment related non-irAEs included anemia, fatigue, bone pain, fever, nausea and vomiting. There was no treatment-related death.
Table 4.
Reported adverse events in overall study population (n = 103 patients totally)
| Adverse events (AE)a | Cutaneous melanoma | Mucosal melanoma | MUP | Total | ||||
|---|---|---|---|---|---|---|---|---|
| No. patients (%) | 83 (100) | 7 (100) | 13 (100) | 103 (100) | ||||
| All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Patients with at least one AE | 79 (95) | 36 (43) | 6 (86) | 4 (57) | 13 (100) | 7 (54) | 98 (95) | 47 (46) |
| Patients with treatment-related AE | 57 (69) | 14 (17) | 3 (43) | 2 (29) | 11 (85) | 4 (31) | 71 (69) | 20 (19) |
| Patients with any irAE | 40 (48) | 12 (15) | 2 (29) | 1 (14) | 10 (77) | 4 (31) | 52 (51) | 17 (17) |
| irDermatitis | 21 (25) | – | 1 (14) | – | 3 (23) | – | 25 (24) | – |
| Pruritus | 8 (10) | – | 1 (14) | – | 2 (15) | – | 11 (11) | – |
| Rash | 8 (10) | – | – | – | 1 (8) | – | 9 (9) | – |
| Erythema multiforme | 4 (5) | – | – | – | – | – | 4 (4) | – |
| Hand-foot-syndrome | 1 (1) | – | – | – | – | – | 1 (1) | – |
| irGastrointestinal disorders | 39 (47) | 15 (18) | 2 (28) | 1 (14) | 8 (62) | 4 (31) | 49 (48) | 20 (20) |
| Colitis | 6 (7) | 4 (5) | – | – | 1 (8) | 1 (8) | 7 (7) | 5 (5) |
| Diarrhea | 25 (30) | 8 (10) | 1 (14) | 1 (14) | 4 (31) | 1 (8) | 30 (29) | 10 (10) |
| GI-perforation | 1 (1) | 1 (1) | – | – | – | – | 1 (1) | 1 (1) |
| Otherb | 7 (9) | 2 (2) | 1 (14) | – | 3 (23) | 2 (15) | 11 (11) | 4 (4) |
| irEndocrine disorders | 5 (6) | 1 (1) | – | – | – | – | 5 (5) | 1 (1) |
| Hypophysitis | 4 (5) | 1 (1) | – | – | – | – | 4 (4) | – |
| Hypothyroidism | 1 (1) | – | – | – | – | – | 1 (1) | – |
| irHepatic disorders | 4 (5) | 1 (1) | – | – | – | – | 4 (4) | 1 (1) |
| Increased ALT | 1 (1) | – | – | – | – | – | 1 (1) | – |
| Increased AST | 1 (1) | – | – | – | – | – | 1 (1) | – |
| Other | 2 (2) | 1 (1) | – | – | – | – | 2 (2) | 1 (1) |
ir immune related, GI gastrointestinal, ALT alanine aminotransferases, AST aspartate aminotransferases, MUP melanoma of unknown primary
aPatients may have had more than one adverse event
bOther gastrointestinal disorders were abdominal pain (n = 6 grade 1/2; n = 3 grade 3/4), constipation (n = 1 grade 1/2) and elevated lipase (n = 1 grade 3/4)
Discussion
This prospective DeCOG phase II trial evaluated the efficacy and safety of ipilimumab in a cohort of 103 patients with 83 pretreated metastatic cutaneous, seven mucosal and 13 occult melanoma. The distribution rate of these clinical subgroups in our trial—considered as representative for a daily routine hospital setting—has been very similar to the rates reported for a named-patient program in Germany with approximately 200 patients [14] [Data not disclosed]. In both multi-center studies, patients with pretreated cutaneous melanoma represented approximately 80 % of all patients; patients with mucosal melanoma (DeCOG: 7 %; expanded access program (EAP) Germany: 5 %) and with occult melanoma (DeCOG: 13 %; EAP Germany: 11 %) were enrolled less frequently. Very similar distribution rates were also reported from large EAPs with 3 mg/kg ipilimumab in Italy, Spain and Australia [15–17] (Additional file 4: Table S4), with rather constant percentages of mucosal melanoma patients (ranging from 7 to 8 %) and occult melanoma patients (6–8 %) enrolled.
The reported OS rate at 12 months of 38 % for patients with cutaneous melanoma is fitting with data from several other studies (range 33–38 %) (Additional file 4: Table S4) enrolling daily clinical routine patients with a high portion of this melanoma subgroup [14–17]; a report on an EAP run in the Netherlands and the UK with cutaneous melanoma patients only also resulted into an 1-year OS rate of 38 % [18]. In a pivotal, randomized phase II dose-ranging study, in which patients with ocular and mucosal melanoma were excluded as well as patients with brain metastases, the OS rate at 12 months for the ipilimumab 3 mg/kg arm of a similar size was 39 % [19]. 2-year survival rates in our study (22 %), in the Dutch-UK expanded access cohort (23 %) [18] and in the pivotal phase II study (24 %) [19] were very similar too: these rates also match with recently published data from a pooled analysis of long-term survival data from approximately 5000 patients included in the ipilimumab pivotal clinical trials and the EAP [20]. This landmark analysis depicts a plateau in the iplilimumab survival curves at around 20 %.
Compared to skin melanoma, primary noncutaneous melanomas show a less favorable outcome when treated with 3 mg/kg ipilimumab. For occult and mucosal melanoma we found lower 1-year survival rates with 27 and 14 %, respectively. Due to the very low patient numbers, the figures for mucosal melanoma are difficult to interpret. However, similar findings were reported from the Australian EAP [17]—the only other study so far reporting OS outcomes for cutaneous versus noncutaneous melanoma treated with 3 mg/kg ipilimumab. Here, the median OS for cutaneous melanoma was twice as high (11.7 months) as for patients with uveal (5.7 months) or mucosal (5.8 months) melanoma. Again, the low number of patients with noncutaneous melanoma resulted into very huge confidence intervals. In the Italian EAP, a median OS of 6.4 months was reported for 71 patients with mucosal melanoma [21]—a value slightly below the 6.8 months reported for the overall patient collective of around 850 patients, 74 % of them with cutaneous melanoma [15]. However, survival data for cutaneous melanoma patients only were not reported from Italy. A retrospective case series from the US similarly found a median OS of 6.4 months for a cohort of 34 mucosal melanoma patients [22]. These estimates all remain below the median OS of 10–11 months, reported for primarily cutaneous melanoma patient collectives in the pivotal phase III trial with highly selected patients and a retrospective long-term-survival landmark analysis [6, 20]. The observed OS difference may be explained by the acknowledged aggressive character of mucosal melanoma; this clinical subgroup represents distinct clinicopathological and molecular features linked with reduced survival rates [10, 21]. For the 13 patients with occult melanoma, data for comparison of efficacy are not available from other studies (Additional file 4: Table S4). Due to the small sample, the inconclusive outcomes in terms of median overall survival and 1-year survival rate do not allow any conclusion, although a better survival outcome for stage IV patients with nodal metastasis of melanoma from an unknown primary (MUP) versus melanoma from a known primary has been reported in a retrospective cohort study [23]. The relative high portion of occult melanoma patients in our initial study cohort, as compared to literature [24, 25], is considered as a selection effect, because such patients usually cannot be included into clinical trials.
In our study, four doses of ipilimumab, the absence of brain metastases, and an ALC ≥1000/µl at week 4 were identified as factors independently associated with a better OS in the 83 patients with cutaneous melanoma. These findings enforce the current level of evidence gained by several studies that the completion of the four-dose-induction phase [14, 26], the absence of brain metastases [14–16], and high ALC counts and/or changes in ALC pharmacodynamics [16–18, 26–29] are predictive for a significant prolongation of survival of ipilimumab-treated patients. Investigations continue to further clarify the role of ALC as an on-treatment pharmacodynamic marker of ipilimumab activity. However, biomarkers to select upfront the right patients for ipilimumab use are still missing. The identification of an immunological biomarker during the development of the anti PD-1 inhibitor nivolumab [30, 31] and the subsequent validation of PD-L1 expression in the course of the pivotal phase III trials [2, 32–34] documents the potential and usefulness of such an approach. Ultimately, melanoma patients are expected to further benefit from a combination of such immunological treatment approaches, administered either sequentially [2, 4, 5] or concomitantly [35]. Furthermore, two PD-1 inhibitors, pembrolizumab [34] and nivolumab [33], as well as the combination of nivolumab and ipilimumab have been shown to improve the progression-free [33, 34] and overall survival [34] compared with ipilimumab in phase 3 clinical trials in patients with metastatic melanoma.
Similar to previous studies of ipilimumab at a dose of 3 mg/kg [6, 36] immune-related dermatological AEs, i.e. pruritus and rash, and immune-related gastrointestinal AEs, i.e. diarrhea and colitis were the most frequent treatment-related adverse events. The rate of grade 3 and 4 treatment-related AEs in patients with cutaneous melanoma were in line with the results of the pivotal phase III trial of ipilimumab [6]. Most of the irAEs were reversible when managed as per protocol-specific treatment guidelines and resolved with systemic glucocorticosteroid therapy. No new and unexpected safety findings were noted except one death with unknown cause was reported and the causal relationship to ipilimumab could not be excluded as per investigator.
Our phase II trial was limited by several factors; (1) the single-arm, non-randomized phase II design, however, at the time of study enrollment, no clear standard therapy for pretreated metastatic melanoma, especially for metastatic mucosal melanoma existed, (2) the small sample sizes of patients with mucosal and occult melanoma, (3) the lack of central review of imaging studies, and (4) the missing classification of tumor assessments according to immune-related response criteria [37].
Conclusions
In conclusion, ipilimumab is a treatment option for patients with advanced cutaneous melanoma seen in daily routine. Given the small number of patients with metastatic mucosal and occult melanoma, it is not possible to determine whether ipilimumab has activity in these melanoma subgroups. The ALC at week 4 appears to be an early biomarker of response and need further confirmation in randomized controlled trials. Immune-related AEs were manageable and reversible in most of the cases.
Authors’ contributions
LZ and DS devised the study concept. DS served as study sponsor and coordinating investigator. All authors were responsible for data acquisition, data assembly and quality control of the data, and reviewed the study report. LZ wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful to patients, their families and staff at the study sites and thank the Alcedis team for statistical support. Editorial and writing assistance was provided by Dr. Markus Hartmann, funded by Bristol-Myers Squibb.
Funding
DS received funding by Bristol Myers-Squibb for the DeCOG-MM-PAL11 trial. Bristol-Myers Squibb study number: CA184-137; http://www.b-ms.de.
Role of the funding source
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors had the final responsibility in submitting the manuscript for publication.
Competing interests
Lisa Zimmer has served as consultant and received speaker’s honoraria and meeting support from Roche, Bristol-Myers Squibb, MSD Sharp & Dohme, Novartis, Merck and GlaxoSmithKline. Thomas K. Eigentler has served in a consultant/advisory role for Bristol-Myers Squibb and has received honoraria from Genentech/Roche, Bristol-Myers Squibb and Amgen. Felix Kieker has served in a consultancy/advisory role for MSD Sharp & Dohme, Brisol-Myers Squibb, Roche, GlaxoSmithKline, Novartis and Amgen. Jan Simon has served in a consultancy/advisory role for Novartis, Roche Pharma and has received support to attend meetings from Merck Sharp & Dohme. Jochen Utikal has served in consultancy/advisory role for Roche and Glaxo SmithKline (GSK). Peter Mohr has served in a consultant/advisory role for MSD Sharp & Dohme (MSD), Leo Pharma, Novartis, GSK, Bristol Myers Squibb, PPD, Hoffmann La Roche, Quintiles. Carola Berking has served in a consultancy/advisory role for Amgen, AstraZeneca, BMS, GSK, MSD, Novartis, and Roche, she has received speaker´s honoraria by BMS, GSK, MSD, Novartis, and Roche and she has received support to attend scientific meetings by BMS, MSD, and Roche. Eckhart Kämpgen received travel expenses and honoraries from Bristol Myers Squibb, fees from GSK and Roche, and served in an advisory role for Beckmann-Coulter. Rudolf Stadler has served as in consultancy/advisory role for Roche and Galderma. Axel Hauschild has served in a consultancy role for Amgen, BMS, Celgene, Eisai, GSK, MedImmune, MelaSciences, Merck Serono, MSD Sharp & Dohme, Novartis, Oncosec, Roche Pharma and received honoraries and trial grants (except from MedImmune) from these corporations. Patrick Terheyden has served in an advisory role for Roche, BMS, Amgen, and GKS with honoraria received from BMS and Roche. Carmen Loquai has served in an advisory role for BMS, Roche, MSD, Novartis, Amgen, receiving honoraria from BMS, Roche, MSD and reimbursement for travel expenses from BMS, Roche, MSD and Novartis. Claus Garbe reports grants and personal fees from BMS during the conduct of the study; personal fees from Amgen, MSD and Novatis, and grants and personal fees from GSK and Roche outside the submitted work. Dirk Schadendorf has served on advisory boards and on speaker´s bureaus of the following companies: Amgen, Boehringer Ingelheim, BMS, Delcath, GSK, Merck-Serono, MSD, Novartis, and Roche; he also served as consultant for BMS, GSK, MSD and Novartis and his institute received research funding from MSD and BMS. None of the remaining authors have any competing interests to disclose.
Additional files
10.1186/s12967-015-0716-5 Consort Diagram.
10.1186/s12967-015-0716-5 Protocol DeCOG –MM-PAL11-Trial.
10.1186/s12967-015-0716-5 Patients with different response pattern in intracranial and extracranial metastases.
10.1186/s12967-015-0716-5 Summarized dataa reporting the efficacy of ipilimumab 3 mg/kg in clinical-practice-settings incl. EAP.
Contributor Information
Lisa Zimmer, Email: lisa.zimmer@uk-essen.de.
Thomas K. Eigentler, Email: thomas.eigentler@med.uni-tuebingen.de
Felix Kiecker, Email: felix.kiecker@charite.de.
Jan Simon, Email: Jan.simon@medizin.uni-leipzig.de.
Jochen Utikal, Email: jochen.utikal@umm.de.
Peter Mohr, Email: peter.mohr@elbekliniken.de.
Carola Berking, Email: carola.berking@med.uni-muenchen.de.
Eckhart Kämpgen, Email: Kaempgen@dermatologikum-berlin.de.
Edgar Dippel, Email: dippele@klilu.de.
Rudolf Stadler, Email: rudolf.stadler@muehlenkreiskliniken.de.
Axel Hauschild, Email: ahauschild@dermatology.uni-kiel.de.
Michael Fluck, Email: michael.fluck@fachklinik-hornheide.de.
Patrick Terheyden, Email: patrick.terheyden@uksh.de.
Rainer Rompel, Email: rompel@klinikum-kassel.de.
Carmen Loquai, Email: Carmen.Loquai@unimedizin-mainz.de.
Zeinab Assi, Email: zeinab.jradi.assi@gmail.com.
Claus Garbe, Email: claus.garbe@uni-tuebingen.de.
Dirk Schadendorf, Email: dirk.schadendorf@uk-essen.de.
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 4.Dummer R, Daud A, Puzanov I, Hamid O, Schadendorf D, Robert C, et al. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. J Transl Med. 2015;13:2062. [Google Scholar]
- 5.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–918. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robert C, Thomas L, Bondarenko I, O’Day S, M DJ, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. [DOI] [PubMed]
- 8.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300–5309. doi: 10.1158/1078-0432.CCR-13-0143. [DOI] [PubMed] [Google Scholar]
- 10.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 11.Zimmer L, Vaubel J, Mohr P, Hauschild A, Utikal J, Simon J, et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS One. 2015;10:e0118564. doi: 10.1371/journal.pone.0118564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Eigentler TK, Schlaak M, Hassel JC, Loquai C, Stoffels I, Gutzmer R, et al. Effectiveness and tolerability of ipilimumab: experiences from 198 patients included in a named-patient program in various daily-practice settings and multiple institutions. J Immunother. 2014;37:374–381. doi: 10.1097/CJI.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 15.Ascierto PA, Simeone E, Sileni VC, Pigozzo J, Maio M, Altomonte M, et al. Clinical experience with ipilimumab 3 mg/kg: real-world efficacy and safety data from an expanded access programme cohort. J Transl Med. 2014;12:116. doi: 10.1186/1479-5876-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berrocal A, Arance A, LopezMartin JA, Soriano V, Munoz E, Alonso L, et al. Ipilimumab for advanced melanoma: experience from the Spanish Expanded Access Program. Melanoma Res. 2014;24:577–583. doi: 10.1097/CMR.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander M, Mellor JD, McArthur G, Kee D. Ipilimumab in pretreated patients with unresectable or metastatic cutaneous, uveal and mucosal melanoma. Med J Aust. 2014;201:49–53. doi: 10.5694/mja13.10448. [DOI] [PubMed] [Google Scholar]
- 18.Kelderman S, Heemskerk B, van Tinteren H, van den Brom RR, Hospers GA, van den Eertwegh AJ, et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother. 2014;63:449–458. doi: 10.1007/s00262-014-1528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 20.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Vecchio M, Di Guardo L, Ascierto PA, Grimaldi AM, Sileni VC, Pigozzo J, et al. Efficacy and safety of ipilimumab 3 mg/kg in patients with pretreated, metastatic, mucosal melanoma. Eur J Cancer. 2014;50:121–127. doi: 10.1016/j.ejca.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Postow MA, Luke JJ, Bluth MJ, Ramaiya N, Panageas KS, Lawrence DP, et al. Ipilimumab for patients with advanced mucosal melanoma. Oncologist. 2013;18:726–732. doi: 10.1634/theoncologist.2012-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CC, Faries MB, Wanek LA, Morton DL. Improved survival for stage IV melanoma from an unknown primary site. J Clin Oncol. 2009;27:3489–3495. doi: 10.1200/JCO.2008.18.9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamposioras K, Pentheroudakis G, Pectasides D, Pavlidis N. Malignant melanoma of unknown primary site. To make the long story short. A systematic review of the literature. Crit Rev Oncol Hematol. 2011;78:112–126. doi: 10.1016/j.critrevonc.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 25.de Waal AC, Aben KK, van Rossum MM, Kiemeney LA. Melanoma of unknown primary origin: a population-based study in the Netherlands. Eur J Cancer. 2013;49:676–683. doi: 10.1016/j.ejca.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24:1697–1703. doi: 10.1093/annonc/mdt027. [DOI] [PubMed] [Google Scholar]
- 27.Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–1775. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilgenhof S, Du Four S, Vandenbroucke F, Everaert H, Salmon I, Lienard D, et al. Single-center experience with ipilimumab in an expanded access program for patients with pretreated advanced melanoma. J Immunother. 2013;36:215–222. doi: 10.1097/CJI.0b013e31828eed39. [DOI] [PubMed] [Google Scholar]
- 29.Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caraco C, Curvietto M, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63:675–683. doi: 10.1007/s00262-014-1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grosso JF, Horak CE, Inzunza D, Cardona DM, Simon JS, Kumar GA, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538) J Clin Oncol. 2013;31(suppl):3016. [Google Scholar]
- 32.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N Engl J Med. 2014;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 33.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 35.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37:499–507. doi: 10.1053/j.seminoncol.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]