

Abstract
There is an urgent need to elucidate the mechanistic links between obesity and colon cancer. Convincing evidence for the role of Lgr5+stem cells in colon tumorigenesis has been established, however, the influence of obesity on stem cell maintenance is unknown. We assessed the effects of high fat (HF) feeding on colonic stem cell maintenance during cancer initiation (AOM induced) and the responsiveness of stem cells to adipokine signaling pathways. The number of colonic GFP+stem cells was significantly higher in the AOM-injected HF group compared to the LF group. The Lgr5+stem cells of the HF fed mice exhibited statistically significant increases in cell proliferation and decreases in apoptosis in response to AOM injection compared to the LF group. Colonic organoid cultures from lean mice treated with an adiponectin receptor agonist exhibited a reduction in Lgr5-GPF+stem cell number and an increase in apoptosis, however this response was diminished in the organoid cultures from obese mice. These results suggest that the responsiveness of colonic stem cells to adiponectin in diet-induced obesity is impaired and may contribute to the stem cell accumulation observed in obesity.
Keywords: Obesity, colon cancer, Lgr5 stem cells, adiponectin
1. Introduction
Colorectal cancer is the 2nd leading cause of cancer mortality in North America [1]and up to 20% of all cancer related deaths may be attributed to obesity [2]. A growing body of data suggest that the relative risk of colon cancer increases in proportion to body mass index tertiles, particularly in men [3-9]. With the rising prevalence of obesity, there is an urgent need to elucidate the mechanistic links between chronic inflammation in adipose tissue and colon cancer risk in obesity. The development of obesity is characterized by excess nutrient delivery to the adipose tissues and an expansion in adipose mass which disrupts the dynamic role of the adipocyte in energy homeostasis, resulting in the alteration of adipose-derived hormones, and development of chronic inflammation [10, 11]. This is significant because studies in both humans and mouse models have provided a clear link between inflammation and cancer [12-14]. The chronic low-grade inflammation associated with obesity may play a key role linking excess adipose tissue, altered adipokine status and the development of colon cancer by promoting tumor development. Diet-induced obese mice exhibit increased numbers of colonic tumors as well as elevated circulating levels of several cytokines and adipokines [15], however the underlying mechanisms responsible for the promotion of colon cancer development in obesity remain to be determined.
The best characterized adipokines relevant to colon cancer are leptin and adiponectin. These two adipokines are primarily produced by adipose tissue, are altered in obesity, and have been shown to play a role in colonic tumorgenesis [16-21]. Unlike most other adipokines, adiponectin is considered anti-inflammatory and is inversely associated with obesity [22, 23]. Epidemiological data suggest that low adiponectin levels are correlated with increased risk of colon cancer [16-18]and supporting evidence for the protective role of adiponectin against colon cancer has been reported both in mice and cell culture studies [20, 24-26]. However, whether specific adipokines directly influence critical target cell populations such as the colonic stem cells, which are implicated in colon tumorigenesis, remains to be determined.
A long-lived pool of rapidly cycling stem cells in the adult mouse small intestines and colon can be identified by the expression of the leucine-rich-repeat-containing G-protein-coupled receptor 5 (Lgr5). Lgr5+ crypt stem cells have been shown to be the cells of origin of intestinal cancer by breeding of genetically modified Apcflox/floxmice and stem-cell-specific Lgr5-EGFP-IRES-creERT2 knockin mice [27]. Both in vivo and in vitro experiments have demonstrated the role of LGR5 in cell proliferation, migration and colony formation [28]. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to effectively prevent colon cancer in humans and rodent models [29, 30]. The chemopreventative mechanism appears to involve the elimination of Lgr5+ stem cells that are inappropriately activated by oncogenic events by inducing apoptosis [31]. There is convincing evidence for the role of Lgr5+ cells in colon tumorigenesis, however, the influence of obesity on stem cell maintenance is unknown. Thus, the objectives of this study were to determine: (1) the effects of high fat feeding on colonic stem cell maintenance during cancer initiation; and (2) the responsiveness of stem cells from obese mice to the activation of adiponectin signaling pathways.
2. Methods & Experimental Design
2.1 Animals and Diet
All experimental procedures adhered to the guidelines approved by Public Health Service and the Institutional Animal Care and Use Committee at Texas A&M University. A total of 20 male Lgr5-EGFP-IRES-creERT2 transgenic mice (3 months of age) were randomized to 2 different experimental groups. Mice were fed either a high fat (HF; 60% kcal from fat; n=10) or low fat (LF; 10% kcal from fat; n=10) diet (Research Diets, New Brunswick, NJ) for 12 weeks. Body weight and food intake were monitored weekly. At the end of 12 weeks on diet, 3 mice per group were injected with saline as a control and 7 mice per group received subcutaneous injections of Azoxymethane (AOM; Sigma-Aldrich) at 15 mg/kg body weight and 13.5 mg/Kg body weight in lean and obese groups, respectively. In order to provide similar amounts of AOM to target tissues, differential doses of AOM for lean and obese mice were used in order to generate equivalent circulating AOM concentrations as previously described [32, 33]. All mice were sacrificed 12 hours after injection.
2.2 Serum Cytokine and Adipokine Profiles
At the time of sacrifice, cardiac blood was collected and allowed to clot at room temperature for 30 min, then centrifuged at 1500 g for 15 min and the serum was stored at minus 80°C. Serum levels of IL-1β, IL-6, IL-17a, leptin, resistin and adiponectin were measured by customized Bio-Plex immunoassays on the Bio-Plex 200 System using Bio-Plex Manager 6.0 software (BioRad). All plasma measurements were analyzed in duplicate.
2.3 Immunohistochemistry
Two hours prior to termination, mice were injected with 30 μg/g body weight of 5-ethynyl-2′ -deoxyuridine (EdU) for analysis of cell proliferation. At the time of sacrifice, the colon was removed and perfused with PBS to remove the contents. A 1 cm section of the distal colon was fixed in 4% PFA in PBS for 4 hours at 4°C, embedded in paraffin, sectioned and used for immunohistochemical studies. The remaining colon was used for isolating colonic crypts that were used for apoptosis assays, cell sorting, and organoid culture experiments as described below.
Fluorescence immunohistochemistry was used to assess markers of cancer initiation, e.g. apoptosis (TUNEL), cell cytokinetics (EdU), and Lgr5 stem cell lineage (GFP) as previously described [34]. GFP was assessed using an anti-GFP primary antibody (Abcam ab6673) followed by an Alexa-488 anti-goat secondary antibody (Jackson Immuno Research cat#705-545-147). Cell proliferation was measured using the Click-IT EdU kit (Invitorgen A-21222) and apoptosis was measured using a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) kit (Trevigen #4810-30). For quantification of immunohistochemical staining, all GFP expressing crypts were counted and the total number of apoptotic and proliferative cells, both GFP+and GFP-, were recorded.
2.4 Organoid Cultures
Colonic crypts were isolated as we have previously described [35, 36]. Briefly, crypts were isolated by incubation with 20 mM EDTA, mechanical disruption and centrifugation. Isolated crypts were resuspended in Matrigel at a density of 15 crypts/ul (BD Biosciences, San Jose, CA), plated onto 24-well plates and maintained in complete medium containing advanced Dulbecco’s modified Eagle’s medium/F12, (ADF; Life Technologies, Grand Island, NY), epidermal growth factor (50 ng/ml; Life Technologies), Noggin (100 ng/ml; Peprotech, Rocky Hill, NJ), R- Spondin (500 ng/ml; Sino Biological, Beijing, China), N2 supplement (1.; Invitrogen), B27 supplement (1X; Life Technologies), N-acetylcysteine (1 μM; Sigma, St Louis, MO) and Wnt-conditioned medium as described previously by Barker et al. [37]. Colonic crypts begin to bud around 2-3 days and on day 3 the organoid cultures from lean and obese Lgr5-GFP mice were treated directly with the recombinant leptin (5-100 ng/ml, BioVision Cat. #4367), adiponectin (5-50 μg/ml, BioVision Cat. #4902), the adiponectin receptor agonist AdipoRon (5-50 uM BioVision Cat. #42565) or dimethyl sulfoxide (DMSO; control vehicle) for 48 hours.
2.5 Flow Cytometry on Organoid Cultures
After 5 days in culture, organoids were harvested by washing with cold PBS then released from the Matrigel by pipetting with cold ADF medium. The dissociated organoids were transferred to a conical tube and residual Matrigel was removed by subsequent PBS washes and centrifugation. Apoptosis was measured using the Dead Cell Apoptosis Kit (Life Tech #V13241) with Annexin V Alexa Fluor 647 conjugate (Life Tech #A23204) following the manufacturer’s instructions. The organoids were incubated with trypsin/DNase to produce a single cell suspension for flow cytometry of apoptotic and GFP positive cells using an Accuri C6 flow cytometer (BD Biosciences).
2.6 Statistics
Investigators were not blinded to the group allocation during the experiment or when assessing the outcome. GraphPad Prism 6.0 and SPSS 14.0 software were used to perform statistical analyses. Body weight, feed intake and serum data were analyzed by the Student’s t-test. Lgr5-GFP+ stem cell number, apoptosis and proliferation data were analyzed using a two-way analysis of variance (ANOVA) with the main effects of diet and depot, and followed, if justified, by testing between-mean differences using Bonferroni’s multiple comparisons test. The D'Agostino & Pearson omnibus normality test was used to test for normality and the Brown-Forsythe test was used for homogeneity of variances. Data sets not normally distributed were log transformed. Differences were considered significant at P <0.05 and all results are reported as mean ± SEM.
3. Results
3.1 High Fat Diet Increased Body Weight and Altered Serum Adipokines
High fat diets are regularly used to induce an obese phenotype, including an increase in body weight, fat mass, adipocyte size and plasma glucose and insulin levels, as well as altered adipokines status [40-42]. We employed a similar high fat dietary approach to induce obesity in the Lgr5-EGFP-IRES-creERT2 transgenic mice. After 12 weeks on diet, the HF group had a significantly higher body weight (p<0.01) compared to the LF group (50 ± 0.8 vs 37± 1.1 grams, respectively). Total food intake was essentially identical between the two groups (240 ± 33 for HF and 240 ± 15 grams for LF). Serum levels of the cytokines IL-1b, IL6 and IL17a did not differ significantly between groups, however, serum leptin levels were increased (p=0.04) and serum adiponectin levels were decreased (p=0.03) in the HF group (Table 1).
Table 1.
Circulating adipokines and cytokines in response to high fat feeding.
| LF | HF | P-values | |
|---|---|---|---|
| IL-1beta (pg/ml) | 1379 ± 866 | 1128 ± 303 | 0.32 |
| IL-17a (pg/ml) | 100 ± 12 | 205 ± 75 | 0.07 |
| IL-6 (ng/ml) | 4.2 ± 1.4 | 1.5 ± 0.8 | 0.13 |
| Resistin (ng/ml) | 90 ± 24 | 83 ± 39 | 0.32 |
| Leptin (ng/ml) | 40 ± 12 | 72 ± 18 | 0.04 |
| Adiponectin (μg/ml) | 8.5 ± 1.6 | 5.3 ± 0.5 | 0.03 |
Data is presented as mean ± SEM for all AOM injected mice (n=7/group). Statistical difference was determined by the Students t-test.
3.2 Effects of High Fat Feeding on Lgr5-GFP+ Stem cell Proliferation and Apoptosis
The total number of cells per crypt did not differ between mice fed HF and LF diets or between AOM-treated and control groups (Figure 1A). We next investigated diet- and treatment-induced differences in distinct Lgr5-GFP+ and non-GFP+cell populations. The number of non-GFP+cells per crypt displayed a pattern similar to total cells per crypt, with neither diet nor AOM treatment exerting a statistically significant effect (Figure 1B). Likewise, the number of Lgr5-GFP+stem cells per crypt did not differ between HF- and LF-fed mice, which had not been injected with AOM (p>0.99), and no statistically significant difference in the number of Lgr5-GFP+stem cells per crypt was observed between AOM-injected LF and non-AOM-injected HF (p=0.09) (Figure 1C).In contrast, AOM-injected HF fed mice had a significantly higher number Lgr5-GFP+stem cells per crypt (p<0.01) compared to AOM-injected LF fed mice (Figure 1C). Additionally, a significant positive correlation (p<0.01) was observed between body weight and Lgr5-GFP+stem cell number per crypt (Figure 1D).
Figure 1.
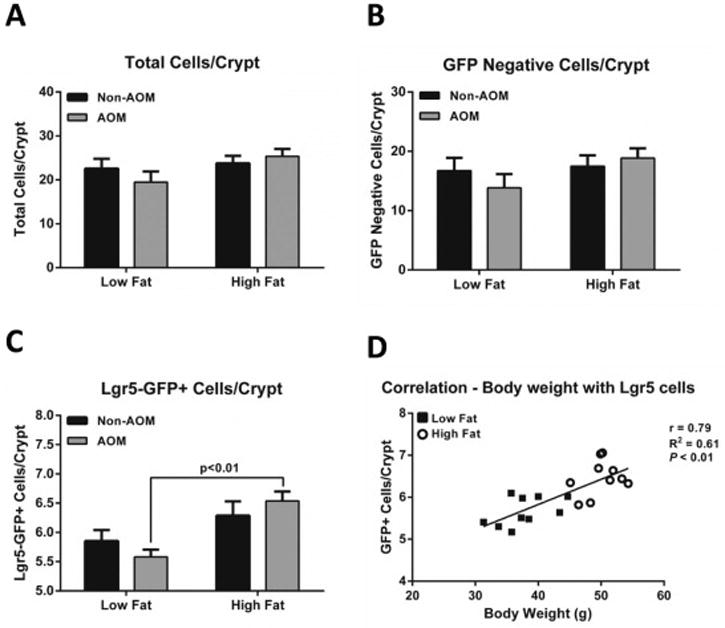
Effect of high fat (HF) feeding and AOM treatment on colonic stem cells. (A) Total cells per crypt, (B) GFP-negative cells per crypt, (C) Lgr5-GFP+cells per crypt, and (D) the correlation between body weight and Lgr5-GFP+ stem cells. Mice were fed a low fat (LF) control diet or a HF diet for 12 wks then injected with AOM or saline. Data represent mean ± SEM (n=3-7 per group). Groups with a p-value <0.05 were considered statistically significant by Bonferroni’s multiple comparisons test following a two-way ANOVA.
To explore the effects of diet-induced obesity on stem cell maintenance in response to AOM induced DNA damage, we examined stem cell apoptosis and cell cytokinetics. Stem cells from LF-fed mice showed a slight reduction in proliferation following AOM injection which was not statistically significant. The percentage of proliferating cells did not differ between diets or between cell types (Lgr5-GFP+and GFP-cells) (Figure 2A and 2B). There were no statistically significant differences in proliferating Lgr5-GFP+cells between non-AOM groups (LF versus HF; p>0.99) or between AOM-injected LF and non-AOM-injected HF groups (p=0.14). Conversely, there was a statistically significant decrease in the number of apoptotic Lgr5-GFP+cells from HF-fed mice compared to the LF group after AOM injection (18% vs 31%, respectively; p<0.01) (Figure 3A). Increased apoptosis was observed in the non-GFP cells in the AOM-injected LF fed mice (9% increase; p<0.01) but not in the HF group. The overall percent induction of apoptosis was much lower in the non-GFP cells compared to the Lgr5-GFP+ cells (Figure 3B). Together, these data suggest that the responsiveness of colonic stem cells to apoptotic signals may be impaired in diet-induced obesity.
Figure 2.
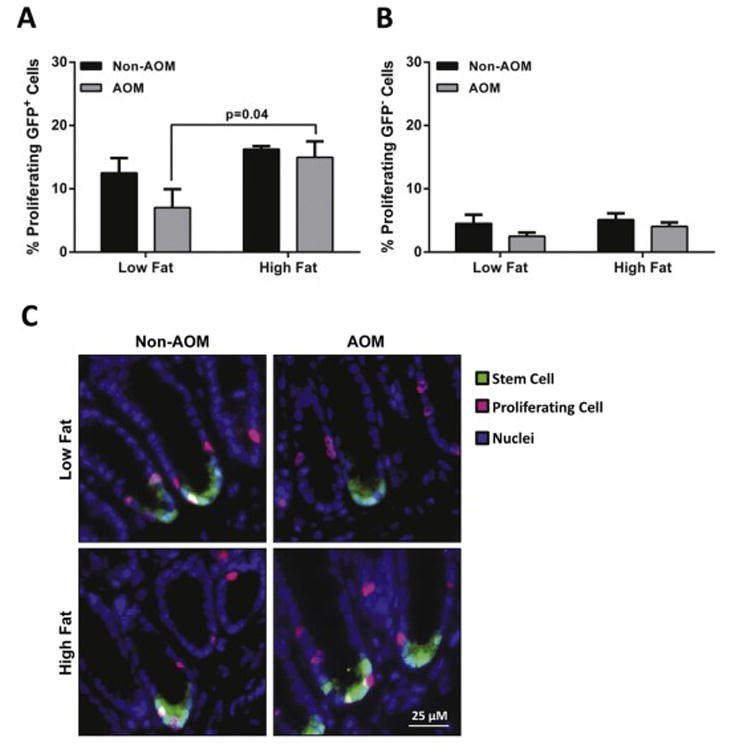
Proliferation of Lgr5-GFP+ and GFP negative cells from obese (HF) mice 12 h after AOM-injection. Colon samples were fixed in 4% PFA and paraffin embedded. Levels of cell proliferation were measured by the EdU Click-It assay. Data are expressed as percentage of EdU-labeled cells relative to the total number of (A) Lgr5-GFP+ stem cells and (B) non-GFP cells. (C) Representative micrographs for EdU—Alexa-647-stained (pink) proliferating cells, Lgr5-GFP+-Alexa-488-stained (green) stem cells and nuclei (blue). Data represent mean ± SEM (n=3-7 per group). Groups with a p-value <0.05 were considered statistically significant by Bonferroni’s multiple comparisons test following a two-way ANOVA.
Figure 3.
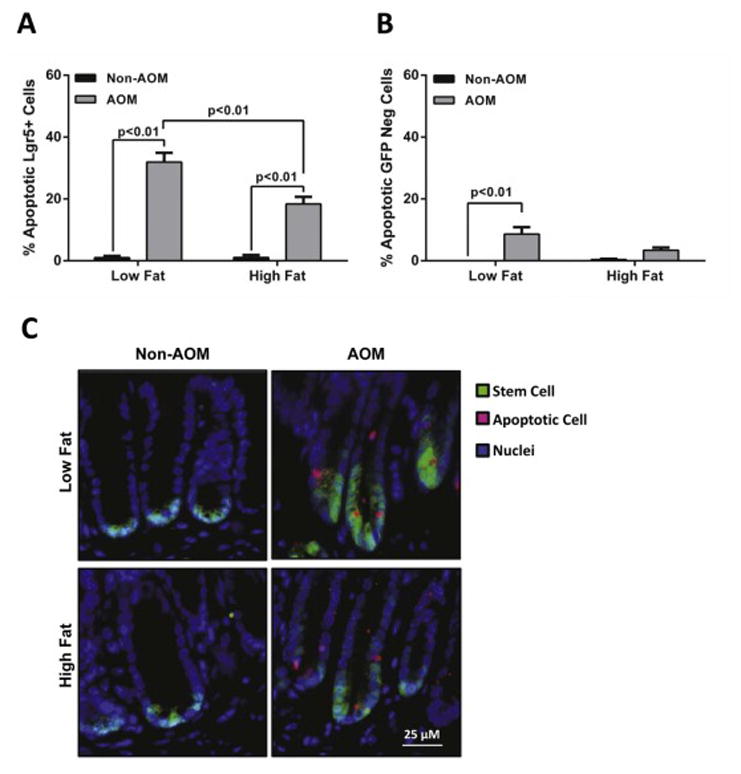
AOM-induced apoptosis is increased in Lgr5-GFP+ colonic stem cells. Colon samples were fixed in 4% PFA and paraffin embedded. Cell apoptosis was measured by the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Data are expressed as the percentage of apoptotic cells relative to the total number of (A) Lgr5-GFP+ stem cells and (B) non-GFP cells. (C) Representative micrographs for TUNEL—Alexa-647-stained (pink) apoptotic cells, Lgr5-GFP+ -Alexa-488-stained (green) stem cells and nuclei (blue). Data represent mean ± SEM (n=3-7 per group). Groups with a p-value <0.05 were considered statistically significant by Bonferroni’s multiple comparisons test following a two-way ANOVA.
3.3 Response of Colonic Organoid Cultures to Adiponectin Receptor Ligation
To determine if altered adipokine status could influence stem cell maintenance, we explored possible adipokine gene induction targets in Lgr5-GFP+cells using RNA-sequencing data from previous experiments [34]. We observed that unlike the leptin receptor or other inflammatory cytokine receptors, both adiponectin receptors (AdipoR1 and AdipoR2) and associated downstream signaling targets (Appl1, AMPK, LKB1) were expressed in colonic Lgr5+stem cells (Figure 4A).Therefore, we proposed that Lgr5+cells, which mark a pool of long-lived cycling intestinal stem cells capable of driving intestinal cancer [27, 28], may be an attractive target for the actions of adiponectin-mediated cues. Results from organoid cultures from lean (standard chow diet) mice indicated that treatment with either recombinant adiponectin or the adiponectin receptor agonist AdipoRon [43] reduced Lgr5-GFP+stem cell number (Figure 4B). In contrast, there was no significant change in Lgr5-GFP+stem cell number when organoid cultures were treated with recombinant leptin (Figure 4B).Therefore, we determined if Lgr5-GFP colonic organoids from obese mice responded differently to activation of the adiponectin signaling pathways.
Figure 4.
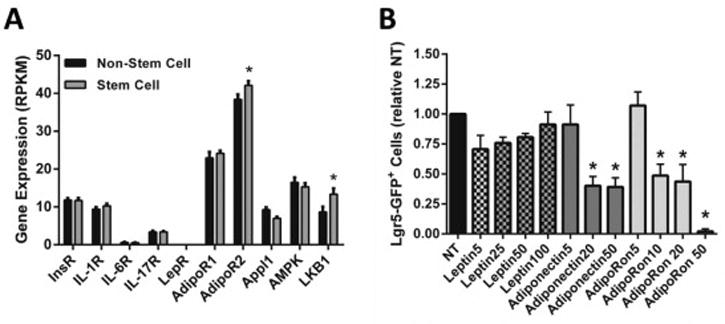
(A) Gene expression of adipokine receptors and several downstream targets identified in stem cells by RNA-sequencing. Data represent mean RPKM values ± SEM (n=20 mice). Statistical difference between stem cell and non-stem cell populations was determined by the Student t-test. (B)Recombinant adiponectin and AdipoRon treatment reduces Lgr5-GFP+ stem cell number in colonic organoid cultures from healthy mice. Colonic crypts were isolated and pooled from regular Lgr5-EGFP-IRES-creER mice (n=2). Organoids were treated with varying doses recombinant leptin, adiponectin or AdipoRon for 48 hours and subsequently analyzed for the percentage of Lgr5-GFP+ stem cells by flow cytometry. Data represent mean ± SEM (treatments were performed in triplicate). Groups with a p-value <0.05 were considered statistically significant by Bonferroni’s multiple comparisons test following a one-way ANOVA.
AdipoRon had very little effect on GFP negative cells in organoid cultures from lean and obese mice (Figure 5A). In contrast, the Lgr5-GFP+stem cell population from lean and obese mice displayed a significant increase in apoptosis in response to AdipoRon treatment (1.6-2.0 fold increase; p≤0.02) (Figure 5A). The overall percent apoptosis was much lower in cultures from obese mice compared to those from lean mice (5.1% NT-HF and 10.1% AdipoRon-HF vs 10.21% NT-LF and 16.21% AdipoRon-LF; p≤0.01) (Figure 5A). Treatment of organoids from lean mice (LF) with AdipoRon resulted in a significant reduction in Lgr5-GPF+ stem cell number. We also observed a 32% reduction in stem cell number in cultures from obese (HF) mice (32% reduction; p=0.05) but the reduction was significantly smaller than that observed in LF mice (63% reduction in LF; p<0.01) (Figure 5B). These differential responses to adiponectin receptor ligation resulted in a significant difference in Lgr5-GFP+ stem cell number between cultures from LF and HF fed mice (p=0.03%) (Figure 5B).
Figure 5.
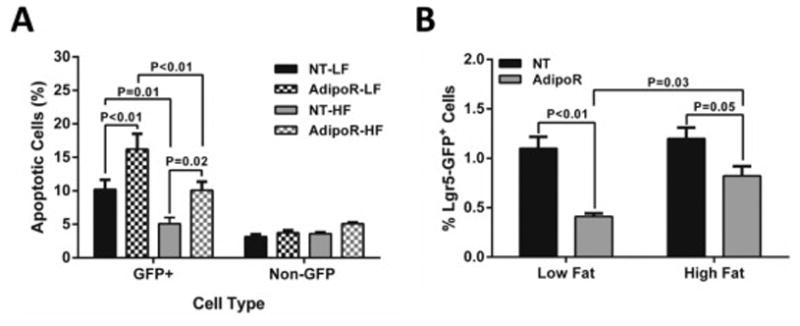
AdipoRon-induced colonic cell apoptosis and reduction of Lgr5-GFP+ stem cell numbers is blunted in organoids cultured from obese (HF) mice. Colonic crypts were isolated from LF (n=3) and HF (n=3) fed Lgr5-EGFP-IRES-creER mice. Organoid cultures from each mouse were treated in triplicate with 10 uM AdipoRon for 48 h and subsequently analyzed for (A) apoptosis and (B) the percentage of Lgr5-GFP+ stem cells by flow cytometry. Data represent mean ± SEM. Groups with a p-value <0.05 were considered statistically significant by Bonferroni’s multiple comparisons test following a two-way ANOVA.
4. Discussion
The association between obesity and colon cancer has been demonstrated in humans and preclinical models, however the underlying mechanisms connecting the two diseases remains largely unknown and only once we understand the workings will be able to prevent or treat obesity-related colon cancer. The current study uses both whole animal and ex vivo culture models to examine the effects of obesity on colonic stem cell proliferation and apoptosis during cancer initiation and the responsiveness of obese colonic stem cells to activation of the adiponectin signaling pathway. We demonstrated the AOM-injected HF group had a significantly higher number of colonic GFP+stem cells compared to the LF group (Figure 1). In response to AOM injection, the HF group also displayed a significant increase in Lgr5-GFP+cell proliferation and a decrease in apoptosis compared to the LF group (Figure 2 & 3). Using the Lgr5-GFP colonic organoid culture system we demonstrated the influence of the adipokine adiponectin on stem cell number and stem cell apoptosis (Figure 5). Collectively the results suggest that the alteration of adiponectin may influence colonic stems in an obese model.
Aberrant crypt foci (ACF), a maker for early development of intestinal cancer, have been reported in diet-induced obese rodent models. Mice fed HF diets ranging from 40-60% fat for 6-77 weeks and injected with AOM exhibit significantly higher ACF than lean control [32, 44-46]. Additionally, an increase in tumor incidence has also been reported with HF feeding in genetic models of intestinal tumorigenesis [46]and tumor-induced AOM models [45, 47]. The AOM model in the current study was based on work of Sikalidis, Fitch, and Fleming [32, 33], providing distinct doses for lean and obese mice to generate equivalent circulating AOM concentrations in order to provide similar amounts of AOM to target tissues. However, other approaches for tumor-induced AOM models have been reported with dose of AOM ranging from 7.5 -15 mg/kg and may produce differing results [20, 32, 45, 47, 48]. Interestingly, mice receiving the HF diet for 8 weeks, then switched back to a low fat diet before the initiation of AOM injections, still displayed an increase in tumor multiplicity despite an attenuation in body weight and fat mass [45], which may suggest more long-term changes occurring in the intestinal epithelium or microbiome. Furthermore, AOM-induced ACF or tumor formation in obese models appears to involve alterations in intestinal epithelial cell proliferation and apoptosis [32, 44, 49]. Mice fed HF diets ranging from 45-60% fat exhibited higher levels of proliferation in colonic epithelial cells [32, 44, 49]and impaired apopototic response to AOM injection [45]compared to LF fed control mice.
Possible mechanisms for the exacerbated AOM effect observed with HF feeding include changes in the functions of the adipose tissue resulting in altered secretion of various regulatory molecules (e.g., adipokines) as well as a more inflammatory phenotype. More severe inflammation is observed in the mesenteric adipose (depot surrounding the intestines) of HF fed (60% fat for 12 weeks) mice compared to LF fed mice [50]. Since obesity is associated with chronic inflammation [50]and colon tumor development is promoted by chronic inflammatory processes [51], the elevation in inflammatory mediators accompanying the obese phenotype likely exacerbates tumorigenesis. Previous data from our lab [41]and others [49]have shown that colonic mucosal injury and inflammation are increased in HF fed mice. In addition, it has been reported that HF fed mice have elevated levels of β-catenin, a key effector of canonical Wnt signaling, as well as expression of c-myc, a downstream target gene.
However, the inflammatory state observed in obesity may not be the only contributor to the increased risk of colon cancer in obese individuals. The excess adipose mass and development of hypertrophic adipocytes is linked to an infiltration of immune cells, further aggravating the inflammatory state, as well as altered secretion and circulating levels of several adipokines and hormones, which may together provide a microenvironment favorable for tumorigenesis [52]. For example, insulin, insulin-like growth factor-1 [53], resistin and visfatin [54]and leptin [21]have all been positively associated with colon cancer, whereas adiponectin levels are significantly lower in colorectal adenoma patients and inversely correlated with the number of adenomas [54].
Adiponectin and leptin are the most widely studied adipokines with respect to colon cancer, however, they may have different role in cancer initiation and progression. In the absence of leptin (ob/ob mice), Endo et al [19] were able to demonstrate an increase in ACF multiplicity (an early indicator of cancer development) with HF feeding, but in long-term experiments the same group also showed that leptin regulated tumor growth, resulting in increased tumor size [19].
Similarly, in a mouse model of intestinal epithelial cell specific leptin receptor knockout, it was documented that both knock out and wild-type had similar number of ACF. ACF number was increased with HF feeding, irrespective of the presence or absence of leptin signaling. But again, long-term tumor studies showed that a local deficiency of the leptin receptor reduced the number of large tumors but not multiplicity [55]. Together these results suggest that leptin may have minimal effects on the early stages of cancer initiation but may contribute more to tumor growth and, thus, there must be other mechanisms contributing to cancer initiation in obesity.
There is a large amount of epidemiological evidence to indicate that low adiponectin levels are inversely correlated with an increased risk of colon cancer, especially in men [16, 18, 56, 57]. Furthermore, well designed studies in both animal and cell culture models further support this link. In several colon cancer cell lines, adiponectin treatment has been shown to increase apoptosis, decrease proliferation and modulate the expression of cell cycle genes through the phosphorylation of ERK1/2, AMPK, LKB1 and S6 ribosomal proteins [25, 26, 58]. In addition, the influence of adiponectin on intestinal carcinogenesis has been examined in both genetic models and chemically induced tumor models. AOM injected adiponectin deficient mice have been reported to have increased colonic epithelial cell proliferation and ACF as well as a significantly higher number of polyps and larger tumor size compared to wild type controls [20, 59, 60]. Similarly, an increased number of intestinal polyps was observed when adiponectin deficient mice (APN-/-) were crossed with Min mice (a genetic model succeptible to intestinal adenomas) as compared to Min mice with normal adiponectin levels (APN+/+). The detrimental effects of adiponectin deficiency on tumorgenesis appear to involve reduced AMPK activation and enhanced mTOR/SK activation [20]. Conversely, treatment with adiponectin reduced the total number of polyps tumor size as well as tumor size and increased apoptosis [26, 60, 61]. Collectively, the previous data indicated that adiponectin suppresses colon tumorigenesis, however, it is unclear whether adiponectin targeted the colonic stem cell niche.
Regional variation in Lgr5 stem cell expression is observed after HF feeding (60% for 16 weeks), with higher expression in the distal region of HF fed mice [62]. HF feeding has also been shown to increase serum and adipose levels of the arachidonic acid-derived PGE2 [63], a prostaglandin that our lab has recently shown to elevate Lgr5-GFP+ cell number and promote cell proliferation in Lgr5 organoid cultures [35]. After consideration of the initial RNA-sequencing data for adipokine receptors, we hypothesized that the inflammatory environment created by high-fat feeding, particularly the impaired secretion of adiponectin, would directly alter colonic stem cell proliferation and apoptosis and promote stem cell expansion and the initiation of colon cancer. Our data indicate that the adiponectin receptor agonist could indeed alter colonic stem cell proliferation and apoptosis in vitro (Figure 5). These data suggest that strategies targeting the adiponectin receptor may be beneficial to stem cell maintenance. However, the biological effects of adiponectin receptor activation with respect to stem cell proliferation and apoptosis in vivo are unknown. Future work should focus on whether activating the adiponectin signaling pathway via the receptor agonist can ameliorate the detrimental effects of obesity on cancer initiation and progression in vivo. This important question could be explored by utilizing the adiponectin receptor agonist AdipoRon in vivo in both acute (12 hours) and chronic (14 weeks) AOM models. Furthermore, this work is limited to the effects of a single adipokine signaling pathway in the organoid culture system, and investigation into other adipose-derived signaling factors that could influence the crosstalk between adiponectin and Lgr5 stem cells is warranted.
In conclusion, our study identifies a potential pathway linking obesity and colon cancer, with the focal point on changes in the secretory profile of adipose tissue during obesity and the impact of adiponectin on colonic stem cell maintenance. During AOM-induced tumor initiation, changes in the colon of obese mice can be characterized by an increase in stem cell number as well as a reduction in stem cell apoptosis and an increase in stem cell proliferation compared to lean mice. Additionally, this work demonstrates that adiponectin is capable of signaling through colonic stem cells. The cells express adiponectin receptors and their activation resulted in a reduction in Lgr5+ stem cell number and increased apoptosis, in the organoid culture system, an important mechanism for preventing tumorgenesis. This research provides evidence to suggest that the adiponectin signaling pathway might represent an attractive therapeutic target for obesity-related colon cancer. Taken together, these experiments provide insight into how altered adipokine secretion observed in obese hypertrophic adipose tissue may modulate the cross talk between adipocytes and colonic stem cells, possibly contributing to the development of colon cancer in obesity.
Highlights.
Colonic stem cell number is significantly higher in obese AOM-injected mice.
Apoptotic response to AOM injection is reduced in high fat fed mice.
Activation of adiponectin signaling pathways induces apoptosis in Lgr5 stem cells.
The responsiveness of colonic stem cells in diet-induced obesity is impaired.
Acknowledgments
The authors would like to thank Dr. Laurie Davidson and Dr. Yang-Yi Fan for technical assistance, and Evelyn Callaway for technical assistance and mouse breeding. V.D., D.N.M., and R.S.C. designed the research; V.D. conducted the research, analyzed data and performed statistical analysis; V.D., and R.S.C. wrote the paper; V.D. and R.S.C. had primary responsibility for final content. All authors have read and approved the final content of the manuscript. Funding from NIH CA129444, CA168312, P30ES023512 and the American Institute for Cancer Research supported this work.
Footnotes
Conflict of interest statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 3.Bassett JK, Severi G, English DR, Baglietto L, Krishnan K, Hopper JL, Giles GG. Body size, weight change, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:2978–2986. doi: 10.1158/1055-9965.EPI-10-0543. [DOI] [PubMed] [Google Scholar]
- 4.Aleksandrova K, Pischon T, Buijsse B, May AM, Peeters PH, Bueno-de-Mesquita HB, Jenab M, Fedirko V, Dahm CC, Siersema PD, Freisling H, Ferrari P, Overvad K, Tjonneland A, Trichopoulou A, Lagiou P, Naska A, Pala V, Mattiello A, Ohlsson B, Jirstrom K, Key TJ, Khaw KT, Riboli E, Boeing H. Adult weight change and risk of colorectal cancer in the European Prospective Investigation into Cancer and Nutrition. Eur J Cancer. 2013;49:3526–3536. doi: 10.1016/j.ejca.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 5.Comstock SS, Hortos K, Kovan B, McCaskey S, Pathak DR, Fenton JI. Adipokines and obesity are associated with colorectal polyps in adult males: a cross-sectional study. PLoS One. 2014;9:e85939. doi: 10.1371/journal.pone.0085939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 7.Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86:556–565. doi: 10.1093/ajcn/86.3.556. [DOI] [PubMed] [Google Scholar]
- 8.Harriss DJ, Atkinson G, George K, Cable NT, Reilly T, Haboubi N, Zwahlen M, Egger M, Renehan AG C-CLEAR group. Lifestyle factors and colorectal cancer risk (1): systematic review and meta-analysis of associations with body mass index. Colorectal Dis. 2009;11:547–563. doi: 10.1111/j.1463-1318.2009.01766.x. [DOI] [PubMed] [Google Scholar]
- 9.Ning Y, Wang L, Giovannucci EL. A quantitative analysis of body mass index and colorectal cancer: findings from 56 observational studies. Obes Rev. 2010;11:19–30. doi: 10.1111/j.1467-789X.2009.00613.x. [DOI] [PubMed] [Google Scholar]
- 10.Karastergiou K, Mohamed-Ali V. The autocrine and paracrine roles of adipokines. Mol Cell Endocrinol. 2010;318:69–78. doi: 10.1016/j.mce.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 11.Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012;249:218–238. doi: 10.1111/j.1600-065X.2012.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 14.Janakiram NB, Rao CV. The role of inflammation in colon cancer. Adv Exp Med Biol. 2014;816:25–52. doi: 10.1007/978-3-0348-0837-8_2. [DOI] [PubMed] [Google Scholar]
- 15.Olivo-Marston SE, Hursting SD, Perkins SN, Schetter A, Khan M, Croce C, Harris CC, Lavigne J. Effects of Calorie Restriction and Diet-Induced Obesity on Murine Colon Carcinogenesis, Growth and Inflammatory Factors, and MicroRNA Expression. PLoS One. 2014;9:e94765. doi: 10.1371/journal.pone.0094765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst. 2005;97:1688–1694. doi: 10.1093/jnci/dji376. [DOI] [PubMed] [Google Scholar]
- 17.Yamaji T, Iwasaki M, Sasazuki S, Tsugane S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Res. 2010;70:5430–5437. doi: 10.1158/0008-5472.CAN-10-0178. [DOI] [PubMed] [Google Scholar]
- 18.Otake S, Takeda H, Fujishima S, Fukui T, Orii T, Sato T, Sasaki Y, Nishise S, Kawata S. Decreased levels of plasma adiponectin associated with increased risk of colorectal cancer. World J Gastroenterol. 2010;16:1252–1257. doi: 10.3748/wjg.v16.i10.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Endo H, Hosono K, Uchiyama T, Sakai E, Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H, Nakajima A. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut. 2011;60:1363–1371. doi: 10.1136/gut.2010.235754. [DOI] [PubMed] [Google Scholar]
- 20.Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, Inamori M, Nakajima N, Watanabe M, Kubota N, Yamauchi T, Kadowaki T, Wada K, Nakagama H, Nakajima A. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut. 2008;57:1531–1538. doi: 10.1136/gut.2008.159293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chia VM, Newcomb PA, Lampe JW, White E, Mandelson MT, McTiernan A, Potter JD. Leptin concentrations, leptin receptor polymorphisms, and colorectal adenoma risk. Cancer Epidemiol Biomarkers Prev. 2007;16:2697–2703. doi: 10.1158/1055-9965.EPI-07-0467. [DOI] [PubMed] [Google Scholar]
- 22.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 23.Ohashi K, Ouchi N, Matsuzawa Y. Anti-inflammatory and anti-atherogenic properties of adiponectin. Biochimie. 2012;94:2137–2142. doi: 10.1016/j.biochi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Fenton JI, Birmingham JM. Adipokine regulation of colon cancer: adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol Carcinog. 2010;49:700–709. doi: 10.1002/mc.20644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, Kim SE, Lee GY, Lee JW, Jung SA, Chung HY, Jeong S, Kim JB. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol. 2010;24:1441–1452. doi: 10.1210/me.2009-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon HS, Liu X, Nagel JM, Chamberland JP, Diakopoulos KN, Brinkoetter MT, Hatziapostolou M, Wu Y, Robson SC, Iliopoulos D, Mantzoros CS. Salutary effects of adiponectin on colon cancer: in vivo and in vitro studies in mice. Gut. 2013;62:561–570. doi: 10.1136/gutjnl-2012-302092. [DOI] [PubMed] [Google Scholar]
- 27.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch D, Barker N, McNeil N, Hu Y, Camps J, McKinnon K, Clevers H, Ried T, Gaiser T. LGR5 positivity defines stem-like cells in colorectal cancer. Carcinogenesis. 2014;35:849–858. doi: 10.1093/carcin/bgt377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 30.Rao CV, Reddy BS. NSAIDs and chemoprevention. Curr Cancer Drug Targets. 2004;4:29–42. doi: 10.2174/1568009043481632. [DOI] [PubMed] [Google Scholar]
- 31.Qiu W, Wang X, Leibowitz B, Liu H, Barker N, Okada H, Oue N, Yasui W, Clevers H, Schoen RE, Yu J, Zhang L. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc Natl Acad Sci U S A. 2010;107:20027–20032. doi: 10.1073/pnas.1010430107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sikalidis AK, Fitch MD, Fleming SE. Diet induced obesity increases the risk of colonic tumorigenesis in mice. Pathol Oncol Res. 2013;19:657–666. doi: 10.1007/s12253-013-9626-0. [DOI] [PubMed] [Google Scholar]
- 33.Sikalidis AK, Fitch MD, Fleming SE. Risk of colonic cancer is not higher in the obese Lep(ob) mouse model compared to lean littermates. Pathol Oncol Res. 2013;19:867–874. doi: 10.1007/s12253-013-9656-7. [DOI] [PubMed] [Google Scholar]
- 34.Davidson LA, Goldsby JS, Callaway ES, Shah MS, Barker N, Chapkin RS. Alteration of colonic stem cell gene signatures during the regenerative response to injury. Biochim Biophys Acta. 2012;1822:1600–1607. doi: 10.1016/j.bbadis.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan YY, Davidson LA, Callaway ES, Goldsby JS, Chapkin RS. Differential effects of 2- and 3-series E-prostaglandins on in vitro expansion of Lgr5+ colonic stem cells. Carcinogenesis. 2014;35:606–612. doi: 10.1093/carcin/bgt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan YY, Davidson LA, Callaway ES, Wright GA, Safe S, Chapkin RS. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am J Physiol Gastrointest Liver Physiol. 2015;309:G1–9. doi: 10.1152/ajpgi.00052.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petro AE, Cotter J, Cooper DA, Peters JC, Surwit SJ, Surwit RS. Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metabolism. 2004;53:454–457. doi: 10.1016/j.metabol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Monk JM, Hou TY, Turk HF, Weeks B, Wu C, McMurray DN, Chapkin RS. Dietary n-3 Polyunsaturated Fatty Acids (PUFA) Decrease Obesity-Associated Th17 Cell-Mediated Inflammation during Colitis. PLoS One. 2012;7:e49739. doi: 10.1371/journal.pone.0049739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffe-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 43.Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, Ogata H, Tokuyama K, Ueki K, Nagano T, Tanaka A, Yokoyama S, Kadowaki T. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–499. doi: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- 44.Endo H, Hosono K, Fujisawa T, Takahashi H, Sugiyama M, Yoneda K, Nozaki Y, Fujita K, Yoneda M, Inamori M, Wada K, Nakagama H, Nakajima A. Involvement of JNK pathway in the promotion of the early stage of colorectal carcinogenesis under high-fat dietary conditions. Gut. 2009;58:1637–1643. doi: 10.1136/gut.2009.183624. [DOI] [PubMed] [Google Scholar]
- 45.Tuominen I, Al-Rabadi L, Stavrakis D, Karagiannides I, Pothoulakis C, Bugni JM. Diet-induced obesity promotes colon tumor development in azoxymethane-treated mice. PLoS One. 2013;8:e60939. doi: 10.1371/journal.pone.0060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day SD, Enos RT, McClellan JL, Steiner JL, Velazquez KT, Murphy EA. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine. 2013;64:454–462. doi: 10.1016/j.cyto.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Huang XF. High fat diet-induced obesity increases the formation of colon polyps induced by azoxymethane in mice. Ann Transl Med. 2015;3:79. doi: 10.3978/j.issn.2305-5839.2015.03.46. 5839.2015.03.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reilly SM, Saltiel AR. Countering inflammatory signals in obesity. Nat Immunol. 2014;15:410–411. doi: 10.1038/ni.2874. [DOI] [PubMed] [Google Scholar]
- 49.Liu Z, Brooks RS, Ciappio ED, Kim SJ, Crott JW, Bennett G, Greenberg AS, Mason JB. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. J Nutr Biochem. 2012;23:1207–1213. doi: 10.1016/j.jnutbio.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paik J, Fierce Y, Treuting PM, Brabb T, Maggio-Price L. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a-/- male mice. J Nutr. 2013;143:1240–1247. doi: 10.3945/jn.113.174615. [DOI] [PubMed] [Google Scholar]
- 51.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez-Hernandez AI, Catalan V, Gomez-Ambrosi J, Rodriguez A, Fruhbeck G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front Endocrinol (Lausanne) 2014;5:65. doi: 10.3389/fendo.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giovannucci E. Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr. 2001;131:3109S–20S. doi: 10.1093/jn/131.11.3109S. [DOI] [PubMed] [Google Scholar]
- 54.Nakajima TE, Yamada Y, Hamano T, Furuta K, Matsuda T, Fujita S, Kato K, Hamaguchi T, Shimada Y. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010;101:1286–1291. doi: 10.1111/j.1349-7006.2010.01518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Higurashi T, Endo H, Uchiyama T, Uchiyama S, Yamada E, Ohkubo H, Sakai E, Takahashi H, Maeda S, Wada K, Natsumeda Y, Hippo Y, Nakajima A, Nakagama H. Conditional knockout of the leptin receptor in the colonic epithelium revealed the local effects of leptin receptor signaling in the progression of colonic tumors in mice. Carcinogenesis. 2014;35:2134–2141. doi: 10.1093/carcin/bgu135. [DOI] [PubMed] [Google Scholar]
- 56.Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev. 2012;33:547–594. doi: 10.1210/er.2011-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aleksandrova K, Boeing H, Jenab M, Bueno-de-Mesquita HB, Jansen E, van Duijnhoven FJ, Fedirko V, Rinaldi S, Romieu I, Riboli E, Romaguera D, Westphal S, Overvad K, Tjonneland A, Boutron-Ruault MC, Clavel-Chapelon F, Kaaks R, Lukanova A, Trichopoulou A, Lagiou P, Trichopoulos D, Agnoli C, Mattiello A, Saieva C, Vineis P, Tumino R, Peeters PH, Arguelles M, Bonet C, Sanchez MJ, Dorronsoro M, Huerta JM, Barricarte A, Palmqvist R, Hallmans G, Khaw KT, Wareham N, Allen NE, Crowe FL, Pischon T. Total and high-molecular weight adiponectin and risk of colorectal cancer: the European Prospective Investigation into Cancer and Nutrition Study. Carcinogenesis. 2012;33:1211–1218. doi: 10.1093/carcin/bgs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams CJ, Mitsiades N, Sozopoulos E, Hsi A, Wolk A, Nifli AP, Tseleni-Balafouta S, Mantzoros CS. Adiponectin receptor expression is elevated in colorectal carcinomas but not in gastrointestinal stromal tumors. Endocr Relat Cancer. 2008;15:289–299. doi: 10.1677/ERC-07-0197. [DOI] [PubMed] [Google Scholar]
- 59.Nishihara T, Baba M, Matsuda M, Inoue M, Nishizawa Y, Fukuhara A, Araki H, Kihara S, Funahashi T, Tamura S, Hayashi N, Iishi H, Shimomura I. Adiponectin deficiency enhances colorectal carcinogenesis and liver tumor formation induced by azoxymethane in mice. World J Gastroenterol. 2008;14:6473–6480. doi: 10.3748/wjg.14.6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mutoh M, Teraoka N, Takasu S, Takahashi M, Onuma K, Yamamoto M, Kubota N, Iseki T, Kadowaki T, Sugimura T, Wakabayashi K. Loss of adiponectin promotes intestinal carcinogenesis in Min and wild-type mice. Gastroenterology. 2011;140:2000–8. 2008.e1–2. doi: 10.1053/j.gastro.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 61.Ishikawa M, Kitayama J, Yamauchi T, Kadowaki T, Maki T, Miyato H, Yamashita H, Nagawa H. Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci. 2007;98:1120–1127. doi: 10.1111/j.1349-7006.2007.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Padidar S, Farquharson AJ, Williams LM, Kearney R, Arthur JR, Drew JE. High-fat diet alters gene expression in the liver and colon: links to increased development of aberrant crypt foci. Dig Dis Sci. 2012;57:1866–1874. doi: 10.1007/s10620-012-2092-9. [DOI] [PubMed] [Google Scholar]
- 63.Iyer A, Lim J, Poudyal H, Reid RC, Suen JY, Webster J, Prins JB, Whitehead JP, Fairlie DP, Brown L. An inhibitor of phospholipase A2 group IIA modulates adipocyte signaling and protects against diet-induced metabolic syndrome in rats. Diabetes. 2012;61:2320–2329. doi: 10.2337/db11-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]