

Abstract
Metabolic reprogramming is a central hallmark of cancer, enabling tumor cells to obtain the macromolecular precursors and energy needed for rapid tumor growth. Understanding how oncogenes coordinate altered signaling with metabolic reprogramming and how cancer cells harness cellular metabolism and its metabolites for their survival may yield new insights into tumor pathogenesis. Here, we review the recently identified central regulatory role for mTORC2, a downstream effector of many cancer‐causing mutations, in metabolic reprogramming and cancer drug resistance in glioblastoma. We further consider the emerging concept that mTORC2 may connect genetics with environmental alterations in brain cancer.
Keywords: drug resistance, glioblastoma, metabolic reprogramming, mTORC2, nutrient
Introduction—Growth Factor Receptor Signaling and Metabolic Reprogramming in Brain Cancer
Metabolic reprogramming is a central hallmark of cancer 13, including the highly lethal primary brain cancer glioblastoma (GBM). Tumor cells convert the majority of the glucose they take up into lactate even when sufficient oxygen is present to support oxidative phosphorylation 32. This adaptation, termed “the Warburg effect,” enables cancer cells to use glucose‐derived carbons to meet both the increased biosynthetic and energy demands imposed by rapid tumor growth. The Warburg effect alone cannot account for the full spectrum of metabolic changes in GBM. GBM cells also catabolize glutamine to support tumor cell proliferation 31 and engage in de novo lipogenesis 6, 12 to support tumor growth. Metabolic reprogramming may also exert some of its most important consequences by globally altering gene transcription and epigenetic landscape 14, 18, including through histone and non‐histone protein acetylation 4, 16, 35, 37.
In cancer, including GBM, metabolic reprogramming is frequently a consequence of upstream mutations in the growth factor receptor–phosphoinositide 3‐kinase (PI3K)–v‐akt murine thymoma viral oncogene (Akt)–mechanistic target of rapamycin (mTOR) signaling network, although the underlying mechanisms are incompletely understood 30, 32. The genomic “portrait” of GBM reveals frequent genetic alterations of the key components of the growth factor receptor‐PI3K–Akt signaling pathway that activate mTOR signaling 3, 24, 25, potentially suggesting a central role for metabolic reprogramming in GBM pathogenesis 5. Receptor tyrosine kinase (RTK) gene amplification and/or mutations are detected in 66% of the primary GBM samples analyzed by The Cancer Genome Atlas (TCGA), with EGFR amplification and/or mutation occurring in 57% of cases 3, 10. In addition, PI3K catalytic and regulatory subunit genetic mutations, and phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene deletion and mutation are common events in GBM 3, resulting in constitutive activation of the PI3K–Akt–mTOR hyperactivation 5.
In this mini‐symposium article, we briefly outline a recent set of discoveries that point to a central role for mTOR complex 2 (mTORC2)‐mediated metabolic reprogramming in GBM pathogenesis. Newly identified molecular mechanisms by which mTORC2 reprograms GBM cells are discussed. In addition, recent work demonstrating that elevated nutrient levels can drive resistance to targeted cancer treatments in GBM via its ability to maintain mTORC2 signaling is highlighted, nominating mTORC2 as a central node integrating altered growth factor receptor signaling with nutrient availability in GBM.
mTORC1 and mTORC2—Essential Components in Metabolic Reprogramming
mTOR is a serine/threonine kinase that exists in two distinct complexes, mTORC1 and mTORC2, which differ in their regulation, function and responsiveness to the allosteric inhibitor, rapamycin 36. mTORC1 contains mTOR kinase in association with Raptor, PRAS40, mLST8, Deptor and Tti1/Tel2 and is a validated cancer drug target 15. mTORC1 links upstream growth factor receptor signaling with downstream protein translation and cell proliferation through its substrates S6K1 and 4E‐BP1. mTORC1 promotes anabolic metabolism downstream of activated PI3K–Akt signaling and in response to amino acid nutrient levels 15, 20. mTORC1 also regulates protein degradation, ribosome biogenesis, glucose, lipid and nucleotide metabolism, and autophagy 15, 36 (Figure 1).
Figure 1.
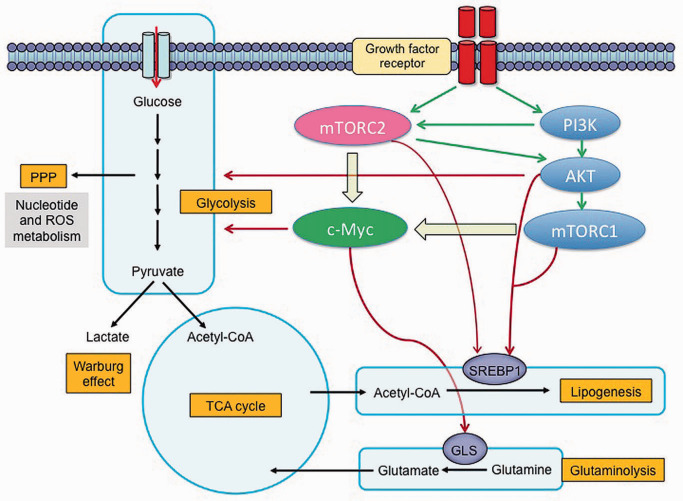
Mechanistic target of rapamycin complex 2 (mTORC2) signaling controls metabolic reprogramming in glioblastoma (GBM): mTORC2 reprograms the glycolytic metabolism, lipid, glutamine, nucleotide and ROS metabolism mainly through Akt and c‐Myc. Akt and c‐Myc promote glycolysis and Warburg effect to generate sufficient energy and macromolecules for rapid tumor growth. mTORC2 activates SREBP1 in an Akt‐dependent and Akt‐independent manner to promote lipogenesis, providing lipids for the synthesis of membrane and signal molecules. mTORC2 also regulates glutaminolysis by activating c‐Myc. GBM with an activated mutant form of EGFR engages c‐Myc signaling at least by two complementary steps, including mTORC1 and mTORC2. GLS = glutaminase; PPP = pentose phosphate pathway; ROS = reactive oxygen species; SREBP = sterol regulatory binding protein; TCA = tricarboxylic acid.
mTORC2 contains mTOR kinase in association with Rictor, mSIN1, Protor1/2, mLST8, Deptor and Tti1/Tel2 15. mTORC2 is activated in response to RTK–PI3K activation and possibly by association with ribosomes 38. mTORC2's most well‐recognized function is the phosphorylation of Akt on Ser473 to promote Akt's maximal activation 27. In GBM, EGFRvIII, the most common epidermal growth factor receptor (EGFR) mutation, and PTEN loss potently activate mTORC2 29. mTORC2 can also activate other protein kinase A/protein kinase G/protein kinase C (AGC) subfamilies, including Akt/PKB, PKCα, and serum and glucocorticoid‐inducible kinase 1 (SGK1). In a Drosophila model of EGFR–PI3K‐driven gliomas, mTORC2, but not Akt or mTORC1, was required for tumor formation 26, suggesting a critical, Akt‐independent role for mTORC2 in GBM pathogenesis. Recent studies suggest that mTORC2 can promote GBM growth and chemotherapy resistance in cancer cells 29, as well as controlling genome stability 28 and tumor metabolism including glycolysis, glutaminolysis, lipogenesis, and nucleotide and reactive oxygen species (ROS) metabolism 22 (Figure 1). These effects appear to occur through Akt‐dependent and Akt‐independent signaling.
Metabolic Reprogramming by mTORC2—C‐Myc, a Master Regulator of mTOR‐Related Metabolism
We recently identified an unexpected Akt‐independent role for mTORC2 in GBM metabolic reprogramming through its ability to control c‐Myc protein level 21. mTORC2 markedly increases glycolysis in GBMs, which is mediated by regulating the intracellular level of c‐Myc, a crucial regulator of the Warburg effect 8. mTORC2 signaling through PKCα, independent of Akt, promotes inactivating phosphorylation of class IIa HDACs 23, leading to constitutive acetylation of FoxO1 and 3, a family of negative regulators of c‐Myc that reduce c‐Myc protein levels through a microRNA‐dependent process 11. Persistent mTORC2 signaling relieves GBM cells of a miR‐34c‐dependent blockade of c‐Myc, potently promoting glycolysis to drive tumor growth. More importantly, activated mTORC2/acetylated FoxO/c‐Myc expression was associated with significantly shorter survival in GBM patients 21.
These results have an intriguing implication; GBM cells are addicted to c‐Myc. c‐Myc is recognized as a central player in cancer, both through c‐Myc's broad transcriptional activities 17 and through its critical role in metabolic reprogramming 8. However, c‐Myc is rarely amplified or mutated in GBM 3, despite its potential importance in GBM pathogenesis. How do the mutations in growth factor receptor signaling pathways, such as EGFR mutations, cooperate with c‐Myc to promote tumorigenesis? Recent work from our laboratory identifies a set of interlacing molecular mechanisms by which EGFRvIII co‐opts c‐Myc to reprogram cellular metabolism and drive tumor proliferation. First, EGFRvIII promotes the oncogenic activity of c‐Myc by inducing mTORC1‐dependent, hnRNPA1‐dependent splicing of the c‐Myc interacting protein MAX, to form the gain of function variant Delta MAX 1. Second, EGFRvIII upregulates c‐Myc protein levels through mTORC2‐dependent acetylation of FoxO1 and 3 21. Taken together, along with a work indicating that Akt can control c‐Myc levels via phosphorylation of FoxO1 and 3 9, 11, these studies identify multiple interlacing complementary mechanisms by which EGFRvIII promotes metabolic reprogramming and tumor cell proliferation via its control of c‐Myc (Figure 1). This tightly integrated, multi‐pronged control mechanism suggests potentially targetable points of therapeutic intervention, but it also raises challenges for molecularly targeted therapies because any of these mechanisms may be sufficient to maintain c‐Myc levels to promote drug resistance. More importantly, failure to inhibit mTORC2, which appears to be harder to target than mTORC1, even with ATP‐competitive kinase inhibitors, may cause tumor cell resistance to PI3K or Akt‐targeted therapies by maintaining elevated levels of c‐Myc.
mTORC2—a Central Player in Drug Resistance
mTORC2 thus appears to be a central player in cancer drug resistance through several pathways. First, mTORC2 may contribute to chemotherapy resistance directly through its activation of Akt. Second, mTORC2 can promote resistance through NF‐κB‐dependent signaling 29. Interestingly, the activation of the NF‐κB pathway in this case is not Akt‐dependent, although Akt has been shown to regulate NF‐κB signaling in other circumstances 2, 7. Third, Akt‐independent mTORC2 signaling has also been shown to promote O‐6‐methylguanine‐DNA methyltransferase (MGMT)‐dependent resistance to one of the most used alkylating chemotherapy in GBM, temozolomide, through N‐Myc downstream regulated gene 1 (NDRG1) 33. These findings suggest that Akt inhibition alone will be insufficient to sensitize tumors to chemotherapy. Fourth, mTORC2 might affect the sensitivity of cancer to chemotherapy as well as radiation through its regulation of genome stability 28, 34. More importantly, TORC2 has also been shown to regulate genome stability in budding yeast 28. To date, the role of mTORC2 in regulating genome instability in human cancer, including GBM, remains unproven and future studies are needed to examine this possibility. Fifth, failure to suppress mTORC2 may promote resistance to PI3K or Akt‐targeted therapies through acetylation of FoxO1 and 3 and maintenance of elevated c‐Myc levels, as described earlier. Thus, mTORC2 may lead to tumor cell resistance to therapy through multiple molecular mechanisms, including metabolic reprogramming.
Emerging Insight—When Genetics and Environments Converge
Cancer metabolic reprogramming is a consequence of upstream mutations in the growth factor receptor–PI3K–Akt–mTOR signaling network, resulting in changes in intracellular nutrient levels. However, do cancer cells also adapt their signaling in response to extracellular nutrient availability? We recently made the surprising discovery that glucose or acetate, two “fuel sources” that are widely available in the brain and readily taken up by tumor cells 19, are required to activate mTORC2 and promote tumor growth 23. Glucose or acetate promoted growth factor receptor signaling through acetyl‐CoA‐dependent acetylation of Rictor, a core component of the mTORC2 signaling complex. Remarkably, in the presence of elevated glucose levels, Rictor acetylation is maintained to form an auto‐activation loop of mTORC2 even when the upstream components of the growth factor receptor signaling pathway are no longer active, thus rendering GBMs resistant to EGFR, PI3K or Akt‐targeted therapies (Figure 2). These results demonstrate that elevated nutrient levels can drive resistance to targeted cancer treatments and nominate mTORC2 as a central node for integrating growth factor signaling with nutrient availability in GBM 23.
Figure 2.
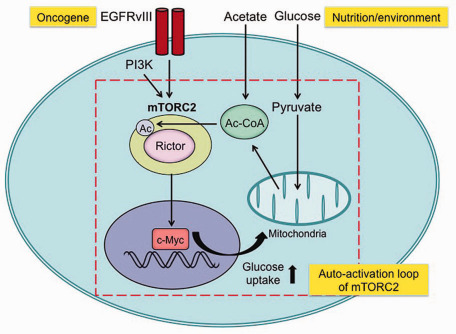
Oncogenic signaling and nutrient environment can interact through mechanistic target of rapamycin complex 2 (mTORC2): mTORC2 forms an auto‐activation loop (i) by promoting glucose/acetate uptake and acetyl‐CoA production through its downstream pathways of c‐Myc 21 and (ii) by an activation of mTORC2 through acetyl‐CoA‐dependent acetylation of Rictor 23. By these mechanisms, glioblastoma (GBM) cells with activated mTORC2 are resistant to targeted therapies toward their upstream stimulators, including epidermal growth factor receptor (EGFR) and phosphoinositide 3‐kinase (PI3K) as well as their downstream effector Akt.
These results have a number of potentially important and unanticipated implications. If sufficient nutrients are present, GBM cells maintain mTORC2 signaling to drive cell proliferation and survival through acetylation‐dependent feedforward activation of mTORC2, suggesting an interplay between oncogenic signaling and the environment. Further, extracellular nutrients can maintain oncogenic downstream growth factor receptor signaling even after tumor cells are treated with inhibitors that target key upstream components of the growth factor receptor signaling system to which they are “addicted,” making the novel prediction that GBM cells, and possibly other cancer types, may use nutrients to escape targeted therapies 23. From a broader perspective, this work begs the question of how lifestyle changes, including diet, can potentially alter tumor cell metabolism, and if so, in what ways. Indeed, there may be more interplay between oncogenic signaling and the environment than previously thought.
Future Perspectives
Cancer cells reprogram their cellular metabolism to meet the biosynthetic and energetic demands imposed by rapid tumor growth and this process appears to be central to GBM pathogenesis. In GBM, mTORC2 appears to be a central node regulating this process, contributing to tumor growth and drug resistance. Signal transduction inhibitors hold the promise of much more effective, much less toxic treatments for GBM patients. However, that promise is unlikely to be realized until the consequences of cancer‐causing mutations on metabolic reprogramming are understood, including the flexible ways in which tumor cells adapt to changing conditions to coordinately maintain the activity of downstream effectors necessary for tumor growth. Unraveling these important questions may point the way toward the more effective targeted cancer treatments, including for patients with GBM.
Acknowledgments
This work was supported by the National Institute for Neurological Diseases and Stroke Grant NS73831; the Defeat GBM Research Collaborative, a subsidiary of National Brain Tumor Society; the National Cancer Institute Grant CA119347; The Ben and Catherine Ivy Foundation; generous donations from the Ziering Family Foundation in memory of Sigi Ziering; and a grant provided by The Ichiro Kanehara Foundation, Uehara Memorial Foundation, The Novartis Foundation (Japan) for the Promotion of Science, and JSPS KAKENHI Grant Number 15K19067. W.K.C. is a Fellow of the National Foundation for Cancer Research. The authors declare no conflict of interest.
References
- 1. Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B et al (2013) EGFR mutation‐induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab 17:1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bai D, Ueno L, Vogt PK (2009) Akt‐mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 125:2863–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325:834–840. [DOI] [PubMed] [Google Scholar]
- 5. Cloughesy TF, Cavenee WK, Mischel PS (2014) Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol 9:1–25. [DOI] [PubMed] [Google Scholar]
- 6. Currie E, Schulze A, Zechner R, Walther TC, Farese RV (2013) Cellular fatty acid metabolism and cancer. Cell Metab 18:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS (2008) Akt‐dependent regulation of NF‐{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev 22:1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dang CV (2013) MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3:a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delpuech O, Griffiths B, East P, Essafi A, Lam EW, Burgering B et al (2007) Induction of Mxi1‐SR alpha by FOXO3a contributes to repression of Myc‐dependent gene expression. Mol Cell Biol 27:4917–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS (2015) Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 15:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M et al (2010) FoxOs enforce a progression checkpoint to constrain mTORC1‐activated renal tumorigenesis. Cancer Cell 18:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H et al (2009) EGFR signaling through an Akt‐SREBP‐1‐dependent, rapamycin‐resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2:ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. [DOI] [PubMed] [Google Scholar]
- 14. Kaelin WG, McKnight SL (2013) Influence of metabolism on epigenetics and disease. Cell 153:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S et al (2014) Akt‐dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE et al (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu C, Thompson CB (2012) Metabolic regulation of epigenetics. Cell Metab 16:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S et al (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159:1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masui K, Cloughesy TF, Mischel PS (2012) Review: molecular pathology in adult high‐grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol 38:271–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T et al (2013) mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c‐Myc. Cell Metab 18:726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masui K, Cavenee WK, Mischel PS (2014) mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab 25:364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Masui K, Tanaka K, Ikegami S, Villa GR, Yang H, Yong WH et al (2015) Glucose‐dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc Natl Acad Sci U S A 112:9406–9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Network CGAR (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Read RD, Cavenee WK, Furnari FB, Thomas JB (2009) A drosophila model for EGFR‐Ras and PI3K‐dependent human glioma. PLoS Genet 5:e1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 28. Shimada K, Filipuzzi I, Stahl M, Helliwell SB, Studer C, Hoepfner D et al (2013) TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol Cell 51:829–839. [DOI] [PubMed] [Google Scholar]
- 29. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B et al (2011) Oncogenic EGFR signaling activates an mTORC2‐NF‐κB pathway that promotes chemotherapy resistance. Cancer Discov 1:524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C et al (2015) Glutamine‐based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7:274ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weiler M, Blaes J, Pusch S, Sahm F, Czabanka M, Luger S et al (2014) mTOR target NDRG1 confers MGMT‐dependent resistance to alkylating chemotherapy. Proc Natl Acad Sci U S A 111:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weisman R, Cohen A, Gasser SM (2014) TORC2—a new player in genome stability. EMBO Mol Med 6:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu SH, Bi JF, Cloughesy T, Cavenee WK, Mischel PS (2014) Emerging function of mTORC2 as a core regulator in glioblastoma: metabolic reprogramming and drug resistance. Cancer Biol Med 11:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiong Y, Guan KL (2012) Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol 198:155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zinzalla V, Stracka D, Oppliger W, Hall MN (2011) Activation of mTORC2 by association with the ribosome. Cell 144:757–768. [DOI] [PubMed] [Google Scholar]