

Abstract
Nε-Thiocarbamoyl-lysine was recently demonstrated by our laboratory to be a potent catalytic mechanism-based SIRT1/2/3 inhibitory warhead, in the current study, among the prepared analogs of Nε-thiocarbamoyl-lysine with its terminal NH2 mono-substituted with alkyl and aryl groups, we found that Nε-methyl-thiocarbamoyl-lysine and Nε-carboxyethyl-thiocarbamoyl-lysine, respectively, also behaved as strong inhibitory warheads against SIRT1/2/3 and SIRT5, typical deacetylases and deacylase in the human sirtuin family, respectively. Moreover, Nε-methyl-thiocarbamoyl-lysine was found in the study to be a ~2.5–18.4-fold stronger SIRT1/2/3 inhibitory warhead than its lead warhead Nε-thiocarbamoyl-lysine.
Keywords: Sirtuin, SIRT1, SIRT2, SIRT3, SIRT5, Deacetylation, Deacylation, Inhibitory warhead, Nε-Methyl-thiocarbamoyl-lysine, Nε-Carboxyethyl-thiocarbamoyl-lysine
Sirtuins are a family of protein Nε-acyl-lysine deacylase enzymes evolutionarily conserved in all the three domains of life.1-4 These enzymes catalyze a type of deacylation reaction in which the Nε-acyl-lysine substrate is condensed with β-NAD+ with the formation of the deacylated product, along with another two products, that is, nicotinamide and 2′-O-AADPR (Fig. 1). The Nε-acyl groups to be removed range from the simple acetyl group to the bulkier succinyl, glutaryl, and myristoyl groups. This enzymatic reaction represents one way of reversing the installation of the Nε-acyl groups on specific lysine side chains on proteins, which can be accomplished enzymatically or non-enzymatically.5,6 Another family of protein Nε-acyl-lysine deacylases are metalloenzymes that harbor a catalytically essential Zn2+ for the hydrolysis of the Nε-acyl-lysine side chain amide functionality.7,8
Figure 1.
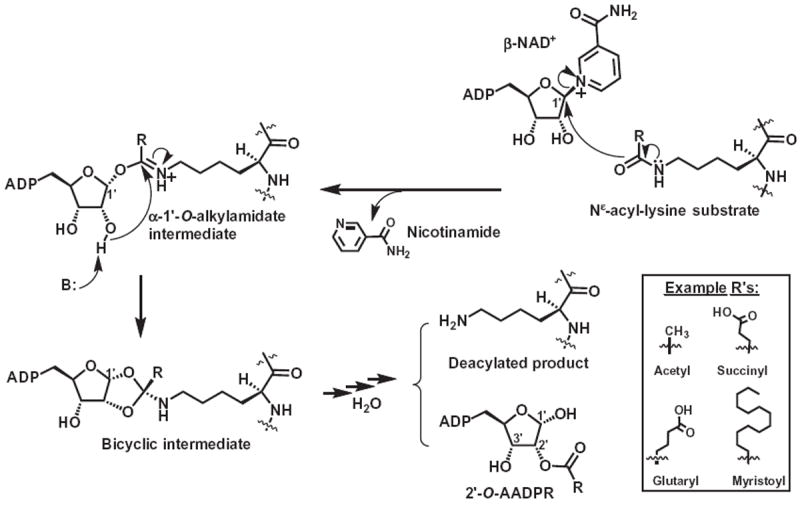
The current version of the chemical mechanism for the sirtuin-catalyzed deacylation of a Nε-acyl-lysine substrate. β-NAD+, β-nicotinamide adenine dinucleotide; 2′-O-AADPR, 2′-O-acyl-ADP-ribose; ADP, adenosine diphosphate; B: refers to a general base.
Since the discovery in 2000 of the protein Nε-acetyl-lysine deacetylase activity for the sirtuin family founding member, that is, the yeast silent information regulator 2 (sir2) protein,9 there have been ever increasing research endeavors on the sirtuincatalyzed deacylation reaction, with three primary focuses being (1) the elucidation of its sophisticated catalytic mechanisms,1,10 (2) the interrogation of its (patho)physiological functions,11-15 and (3) the exploration of the therapeutic potentials via targeting this enzymatic reaction.16,17 The ultimate goal of these endeavors would be to develop novel therapeutic agents via targeting (inhibiting or activating) the sirtuin-catalyzed deacylation reaction. Therefore, there has been an active research on developing chemical modulators (inhibitors and activators) for this enzymatic reaction.1,16,18-21 It is worth noting that such chemical entities would also be valuable chemical biological or pharmacological research tools.
Among the currently existing sirtuin chemical modulators, the catalytic mechanism-based modulators, mostly inhibitors, represent a class of intriguing double-edged compounds in that they have also been instrumental in helping dissect the chemical mechanism of the sirtuin deacylation reaction (Fig. 1).1,18,22 The current catalytic mechanism-based sirtuin inhibitors are primarily based on the Nε-thioacyl-lysine inhibitory warheads,1,18,22,23 with Nε-thioacetyl-lysine being the first such warhead that our laboratory and that of Denu developed during 2006–2007.24,25 In order to address the concern over a thioamide compound’s potential cellular toxicity (especially the hepatotoxicity) following its metabolic S-oxidation and activation thereafter,26-30 our laboratory previously developed Nε-thiocarbamoyl-lysine as a potent and general SIRT1/2/3 inhibitory warhead, with its catalytic mechanism-based nature directly demonstrated with compound 1 and SIRT1, a typical deacetylase in the human sirtuin family (Fig. 2).31 It was found in our study that SIRT1 was able to process compound 1 as a substrate yet with the formation of the depicted α-1′-S-alkylamidate intermediate which was apparently longer-lived so that it was detectable with mass spectrometry. In order to improve its inhibitory potency and to extend this design concept to other human sirtuin family members, in the current study, we prepared seven analogs of Nε-thiocarbamoyl-lysine with its terminal NH2 mono-substituted with alkyl and aryl groups (Figs. 3 and 4). Among these analogs, Nε-methyl-thiocarbamoyl-lysine (the central residue in compound 3, Fig. 3) was found to be also a strong SIRT1/2/3 inhibitory warhead. Moreover, Nε-carboxyethyl-thiocarbamoyl-lysine (the central residue in compound 8, Fig. 4) was found to be a potent inhibitory warhead against SIRT5, a typical deacylase in the human sirtuin family. The addition of these novel thioureabased sirtuin inhibitory warheads into the arsenal would facilitate the development of advanced members (being both potent and safe) in the family of the catalytic mechanism-based inhibitors which have become a major type of the inhibitors for the sirtuincatalyzed deacylation reaction.
Figure 2.
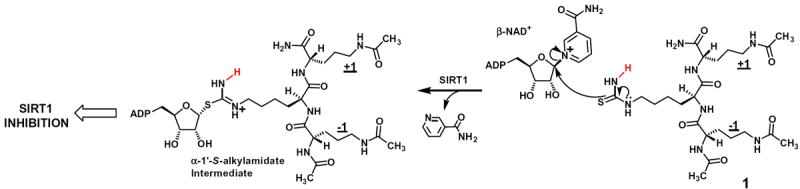
The catalytic mechanism-based SIRT1 inhibition by the tripeptidic compound 1 that harbors the lead inhibitory warhead Nε-thiocarbamoyl-lysine at its central position. SIRT1 was found previously in our laboratory to be able to process compound 1 as a substrate yet with the formation of the longer-lived α-1′-S-alkylamidate intermediate.31 Note: the Nε-thiocarbamoyl-lysine-containing compound 1 has Nε-acetyl-ornithine at both −1 and +1 positions.
Figure 3.
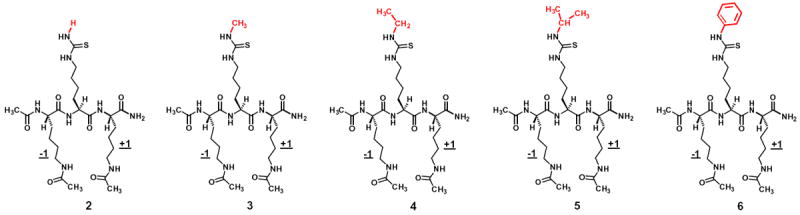
The chemical structures of compound 2 and the initial four analogs of Nε-thiocarbamoyl-lysine embedded in the same tripeptidic scaffold as the central residue. The side chain terminal NH-substituents were colored in red. These analogs were designed to assess their SIRT1/2/3 inhibitory power. Note: the Nε-thiocarbamoyl-lysine-containing compound 2 has Nε-acetyl-lysine at both −1 and +1 positions.
Figure 4.
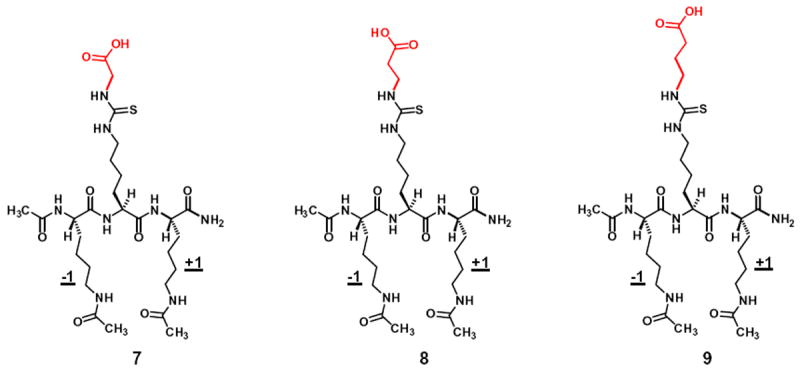
The chemical structures of the three further analogs of Nε-thiocarbamoyl-lysine with their side chain terminal NH-substituents (colored in red) all ending with carboxyl. These analogs were embedded as the central residue in the same tripeptidic scaffold as that in Figure 3, and were designed to primarily assess their SIRT5 inhibitory power.
Our initial effort in the current study was to improve the inhibitory power of the SIRT1/2/3 warhead Nε-thiocarbamoyl-lysine via introducing alkyl and aryl mono-substituents at its terminal NH2. The rationale behind this was the previous demonstration that the active sites of those sirtuins with a robust Nε-acetyl-lysine deacetylase activity were also able to accommodate the Nε-acyl groups bulkier than acetyl, such as propionyl, butyryl, and even long-chain fatty acyl groups like myristoyl.1,18,32 However, unlike the lysine Nε-deacylation of the short-chain fatty acyl groups, that of the long-chain fatty acyl groups seems to be a shared feature of many sirtuins including both the deacetylase members (e.g., SIRT1-3) and the deacylase members (e.g., SIRT5 and SIRT6) in the human sirtuin family. Therefore, the mono-substituents at the terminal NH2 of Nε-thiocarbamoyl-lysine would not include long-chain fatty acyl groups, instead, methyl and ethyl groups (simple alkyl chain homologation), isopropyl group (alkyl chain branching), and phenyl group (an aryl ring) were employed.
To assess the inhibitory power of the newly designed analogs of Nε-thiocarbamoyl-lysine, they were incorporated as the central residue into a tripeptidic scaffold with Nε-acetyl-lysine at both of its –1 and +1 positions (Fig. 3). This scaffold is one of the two proteolytically stable and cell permeable scaffolds that our laboratory discovered and has been using over the past few years when evaluating the performance of a sirtuin inhibitory warhead, which also includes a similar tripeptidic scaffold but with Nδ-acetyl-ornithine at both −1 and +1 positions (the one present in compound 1).18 Because it was found previously in our laboratory that the diacetyl-lysine scaffold was able to confer a stronger SIRT1 inhibition than the di-acetyl-ornithine scaffold,33 we opt to use the former scaffold in the current study. Accordingly, for a direct sirtuin inhibitory potency comparison with analogs 3–6, we also prepared the analog of compound 1 with Nε-acetyl-lysine at both −1 and +1 positions (i.e., compound 2, Fig. 3).
The tripeptidic compounds 2–6 (Fig. 3) were prepared according to Schemes 1 and 2. They were isolated from the respective reaction mixtures by semi-preparative reversed-phase high pressure liquid chromatography (RP-HPLC), and were shown to be >95% pure based on analytic RP-HPLC analysis. Their identities were confirmed by high-resolution mass spectrometry (HRMS) analysis (Table 1).
Scheme 1.
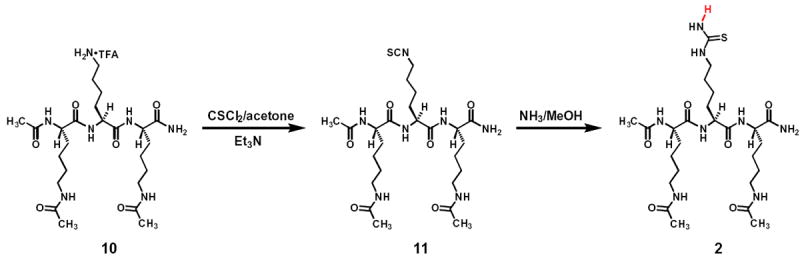
The synthetic scheme for compound 2. The tripeptidic compound 10 was obtained from China Peptides Co., Ltd via a custom synthesis order; its purity was ≥ 98% and its identity was confirmed by ESI-MS analysis.
Scheme 2.
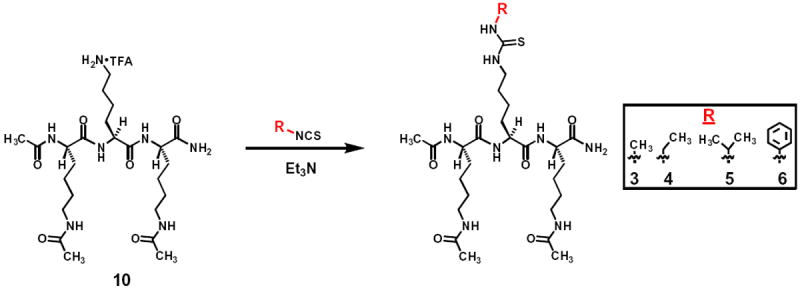
The synthetic scheme for compounds 3–6.
Table 1.
HRMS analysis of compounds 2–9a
| Compound | Ionic formula | Calculated m/z | Observed m/z |
|---|---|---|---|
| 2 | [C25H47N8O6S]+ | 587.3339 | 587.3326 |
| 3 | [C26H48N8O6SNa]+ | 623.3310 | 623.3285 |
| 4 | [C27H50N8O6SNa]+ | 637.3466 | 637.3476 |
| 5 | [C28H52N8O6SNa]+ | 651.3623 | 651.3591 |
| 6 | [C31H50N8O6SNa]+ | 685.3466 | 685.3460 |
| 7 | [C27H48N8O8SNa]+ | 667.3208 | 667.3187 |
| 8 | [C28H50N8O8SNa]+ | 681.3365 | 681.3377 |
| 9 | [C29H52N8O8SNa]+ | 695.3521 | 695.3530 |
All the compounds were measured with the positive mode of electro-spray ionization (ESI).
When compound 2 was assessed for its SIRT1/2/3 inhibitory potencies, as expected, it exhibited a ~1.8-fold stronger SIRT1 inhibition than compound 1 (Table 2). Moreover, a ~5.2-fold and a ~10.6-fold stronger inhibition, respectively, against SIRT2 and SIRT3 were also observed for 2 than 1. When compounds 3–6 were evaluated for their inhibitory potencies against SIRT1, the three Nε-thiocarbamoyl-lysine analogs, respectively, presented by compounds 4–6 (i.e., Nε-ethyl-, Nε-isopropyl-, and Nε-phenylthiocarbamoyl-lysine) seemed to be about 1.3–2.4-fold weaker SIRT1 inhibitory warheads than Nε-thiocarbamoyl-lysine. However, Nε-methyl-thiocarbamoyl-lysine presented by compound 3 was found to be an about 8.9-fold stronger SIRT1 inhibitory warhead than Nε-thiocarbamoyl-lysine (Table 2). Therefore, compound 3 was further assessed for its inhibitory potencies against SIRT2 and SIRT3. As can be seen in Table 2, Nε-methyl-thiocarbamoyllysine seemed to be an ~18.4-fold and an ~2.5-fold stronger SIRT2 and SIRT3 inhibitory warhead, respectively, than Nε-thiocarbamoyl-lysine. Of note, the above sirtuin inhibitory power differences were inferred from a comparison of the sirtuin inhibitory potencies of analogs 3–6 with that of compound 2.
Table 2.
SIRT1/2/3 inhibition by compounds 2–6a
| Compound | IC50 (μM)
|
||
|---|---|---|---|
| SIRT1 | SIRT2 | SIRT3 | |
| 1b | 89.5 ± 16.1 | 159.1 ± 32.8 | 57.2 ± 17.6 |
| 2 | 49.9 ± 3.68 | 30.4 ± 3.6 | 5.4 ± 0.5 |
| 3 | 5.6 ± 3.4 | 1.65 ± 0.64 | 2.2 ± 0.07 |
| 4 | 83.4 ± 47.4 | N.D.c | N.D. |
| 5 | 117.9 ± 75.6 | N.D. | N.D. |
| 6 | 62.9 ± 5.7 | N.D. | N.D. |
While the sirtuin inhibition assay details are fully described in ‘Supplementary material’, the following is the information on the substrate concentration, assay time, and sirtuin concentration, the three key assay parameters employed. [β-NAD+] used: 0.5 mM for the SIRT1 and SIRT2 assays, 3.5 mM for the SIRT3 assay. [Peptide substrate] used: 0.3 mM, 0.39 mM, and 0.105 mM of H2N-HK-(Nε-acetyl-lysine)-LM-COOH for the SIRT1, SIRT2, and SIRT3 assay, respectively. [Sirtuin] used: His6-SIRT1 or GST-SIRT1, 320 nM; His6-SIRT2, 297 nM; His6-SIRT3, 369 nM. Enzymatic reaction times used: 10 min for the SIRT1 and SIRT2 assays, 8 min for the SIRT3 assay.
The SIRT1/2/3 inhibitory data for compound 1 were taken from Ref. 31, which were acquired under the same assay condition as that employed in the current study.
N.D., not determined in the current study.
Since some thiourea compounds were previously reported to be effective scavengers of the superoxide radical anion ( ),34 we were also interested in determining the anti-oxidant potential of Nε-methyl-thiocarbamoyl-lysine whose side chain harbors a thiourea functionality. In the experiment, compound 3 was assessed side-by-side with a Nε-thioacetyl-lysine-containing compound with the same tripeptidic scaffold (shown in Fig. 5). As depicted in Figure 5, while this latter compound did not exhibit a significant anti-oxidant activity against the superoxide radical anion under our experimental condition, with only a ~6% of inhibition at 200 μM of the compound; 3 exhibited a much more significant anti-oxidant activity under the same experimental condition, with a 50% of inhibition at 200 μM of the compound.
Figure 5.
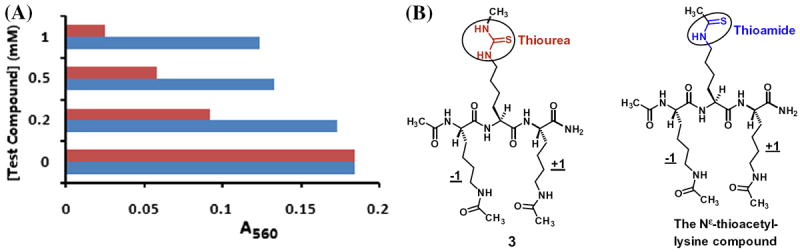
The superoxide radical anion scavenging activities of compound 3 and the depicted Nε-thioacetyl-lysine compound. (A) The bar graph indicating the stronger anti-oxidant activity of compound 3 than the Nε-thioacetyl-lysine compound
 , compound 3;
, compound 3;
 , Nε-thioacetyl-lysine compound. See ‘Supplementary material’ for experimental details. (B) A structural comparison of compound 3 and the Nε-thioacetyl-lysine compound.
, Nε-thioacetyl-lysine compound. See ‘Supplementary material’ for experimental details. (B) A structural comparison of compound 3 and the Nε-thioacetyl-lysine compound.
Compound 3 was also subjected to a pronase digestion assay to assess its proteolytical stability. As shown in Figure 6, as compared to the linear peptide control, compound 3 was found to be much more proteolytically stable. Specifically, while the control peptide was degraded almost completely within 3 min of incubation with pronase, there was still ~88% remaining for compound 3 even after 60 min of incubation with pronase under the same experimental condition. Of note, pronase was employed in this experiment because it has a very broad substrate specificity due to the presence in it of a variety of different types of the proteases/peptidases.35
Figure 6.
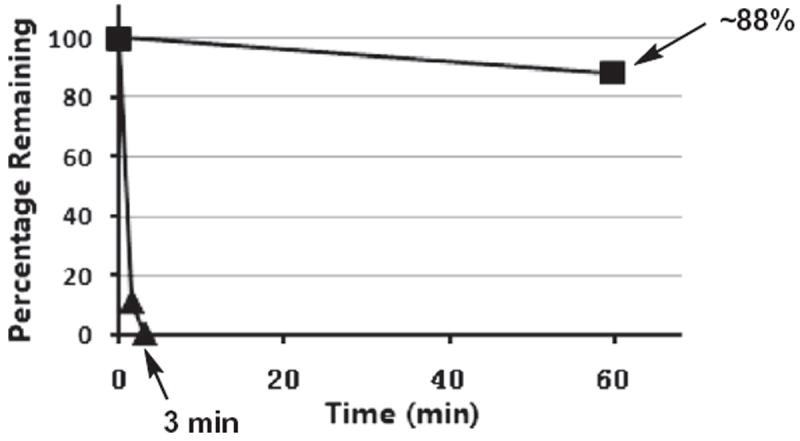
The proteolytic stability of 3, as assessed by the pronase digestion assay. ■, the degradation profile of 3; ▲, the degradation profile of the control: H2N-HK-(Nε-acetyl-lysine)-LM-COOH.
Compound 3 was further examined for its ability to inhibit intracellular SIRT1. For this purpose, a convenient Western blot analysis33 that our laboratory established and has been using in the past few years was employed. As shown in Figure 7, treating human colon cancer HCT116 cells with compound 3 at increasing concentrations (0–250 μM) was able to bring about a dose-dependent increase of the p53 protein acetylation at K382, a native deacetylation site of SIRT1. In Figure 7, the immunoreactivity of the total p53 protein blotted with a total-p53 antibody was also included to exclude the possibility that the observed p53 K382 acetylation level increase was due to an increase in the total p53 protein level.
Figure 7.
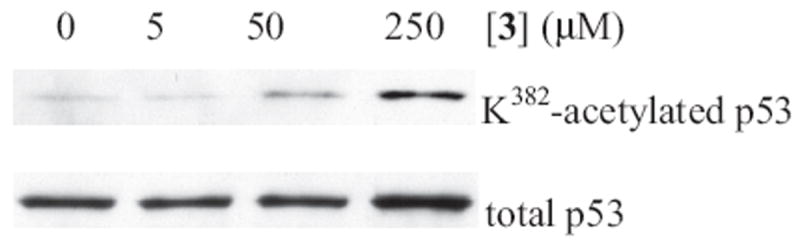
The cell permeability of 3, as assessed by the Western blot analysis of the p53 protein Lys382 acetylation level change in HCT116 human colon cancer cells treated with 3.
Due to the catalytic mechanism (especially the chemical mechanism) conservation for the sirtuin-catalyzed deacylation (including deacetylation)1,10,18 and the demonstrated (with SIRT1) catalytic mechanism-based nature for the lead warhead Nε-thiocarbamoyl-lysine,31 we were interested in extending the design of the thiourea-based sirtuin inhibitory warhead to other members of the human sirtuin family, especially those possessing a strong deacylase activity but a weak deacetylase activity. For this purpose, we picked the typical deacylase SIRT5. Three Nε-carboxyalkyl-thiocarbamoyl-lysines, respectively, embedded in compounds 7, 8, and 9 as the central residue (Fig. 4) were designed based on the previous observations that SIRT5 possessed stronger demalonylase, desuccinylase, and deglutarylase activities than its deacetylase activity.1,3 Of note, compounds 7–9 have the same di-acetyl-lysine tripeptidic scaffold as that in compounds 2–6.
The designed tripeptidic compounds 7–9 were synthesized according to Scheme 3, isolated from reaction mixtures by semipreparative RP-HPLC, and were also shown to be >95% pure based on analytic RP-HPLC analysis. The identities of the compounds 7-9 isolated were also confirmed by HRMS analysis (Table 1).
Scheme 3.
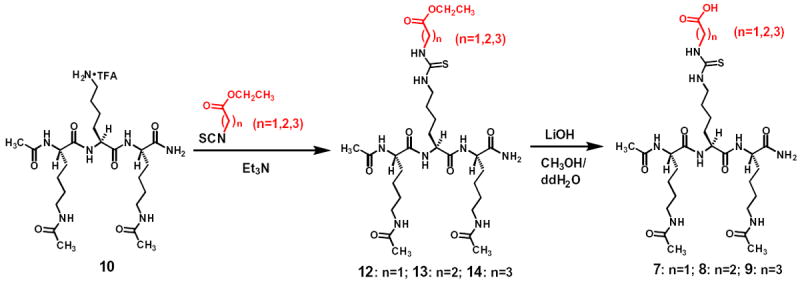
The synthetic scheme for compounds 7–9.
When compounds 7–9 were evaluated with the human SIRT5, compound 8 was found to be a significantly stronger SIRT5 inhibitor than compounds 7 and 9 (Table 3). Therefore, the central residue of compound 8, that is, Nε-carboxyethyl-thiocarbamoyl-lysine, would represent a potent inhibitory warhead against SIRT5. It should be pointed out that the intermediate 13 during the synthesis of compound 8 (Scheme 3), that is, the ethyl ester of 8, was found to be a weak SIRT5 inhibitor with an IC50 value greater than 100 μM. This finding reinforced the notion that the free terminal carboxyl in a Nε-succinyl-lysine substrate is necessary for its tight binding at SIRT5 active site via promoting a salt bridge formation with a catalytically essential arginine side chain.36
Table 3.
Sirtuin inhibitory potencies of compounds 7–9a
| Compound | IC50 (μM)
|
||
|---|---|---|---|
| SIRT5 | SIRT6 | SIRT1 | |
| 7 | > 100 | — | — |
| 8 | 5.0 ± 1.9 | ~2400 | > 100 |
| 9 | > 100 | — | — |
While the sirtuin inhibition assay details are fully described in ‘Supplementary material’, the following is the information on the substrate concentration, assay time, and sirtuin concentration, the three key assay parameters employed. [β-NAD+] used: 0.5 mM for the SIRT1 assay, 0.8 mM for the SIRT5 assay, 0.2 mM for the SIRT6 assay. [Peptide substrate] used: 0.3 mMof H2N-HK-(Nε-acetyl-lysine)-LM-COOH for the SIRT1 assay, 0.88 mM of CH3CONH-AR-(Nε-succinyl-lysine)-ST-CONH2 for the SIRT5 assay, 0.02 mM of H2N-EALPK-(Nε-myristoyl-lysine)-TGGPQ-CONH2 for the SIRT6 assay. [Sirtuin] used: His6-SIRT1 or GST-SIRT1, 320 nM; GST-SIRT5, 370 nM; His6-SIRT6, 313 nM. Enzymatic reaction times used: 10 min for the SIRT1 assay, 5 min for the SIRT5 assay, 12 min for the SIRT6 assay.
Since SIRT6 was also recently found to possess a stronger deacylase (demyristoylase) activity than its ability to catalyze deacetylation,1,3 we were interested in examining whether Nε-carboxyethyl-thiocarbamoyl-lysine could also be an inhibitory warhead against SIRT6 which is another known deacylase member in the human sirtuin family. However, in contrast to its strong inhibition against SIRT5, compound 8 was found to be a weak SIRT6 inhibitor with an IC50 value around 2.4 mM (Table 3). Moreover, compound 8 was also found to be a weak inhibitor against SIRT1 with an IC50 value greater than 100 μM (Table 3). These findings are consistent with the past observation that the succinyl removal from Nε-succinyl-lysine that Nε-carboxyethylthiocarbamoyl-lysine mimics is unique to SIRT5 as compared to SIRT6 and the canonical deacetylase members (SIRT1/2/3) in the human sirtuin family.32
To summarize, in the current study, built upon the lead sirtuin inhibitory warhead Nε-thiocarbamoyl-lysine, we identified Nε-methyl-thiocarbamoyl-lysine as a novel thiourea-based SIRT1/2/3 inhibitory warhead that is ~2.5–18.4-fold stronger than Nε-thiocarbamoyl-lysine and possesses a significant anti-oxidant activity against the superoxide radical anion. We have also identified Nε-carboxyethyl-thiocarbamoyl-lysine as a strong and selective (vs SIRT6 and SIRT1) thiourea-based SIRT5 inhibitory warhead.
The availability of these novel warheads would broaden the horizon in developing more effective inhibitors against the therapeutically important sirtuin-catalyzed deacylation reaction as potential therapeutic agents. In addition to being still potent, the class of the thiourea-based warheads would potentially be able to circumvent the cytotoxicity problem associated with the use of the currently most potent warheads Nε-thioacyl-lysines, rendering the class of the sirtuin inhibitors harboring the thiourea-based warheads potentially safer future sirtuin-inhibiting therapeutics. Moreover, the observed strong anti-oxidant activity of the Nε-methyl-thiocarbamoyl-lysine-containing compound 3 in the current study suggested that the thiourea-based sirtuin inhibitors would be protective toward normal cells; however, in the meantime, this anti-oxidant activity could possess an anti-cancer potential according to the literature precedents.37,38
Supplementary Material
Acknowledgments
We greatly appreciate the financial support to this work from the following: the National Natural Science Foundation of China (Grant No: 21272094), the Jiangsu provincial specially appointed professorship, the Jiangsu provincial ‘innovation and venture talents’ award plan, and Jiangsu University.
Abbreviations
- β-NAD+
β-nicotinamide adenine dinucleotide
- 2′-O-AADPR
2′-O-acyl-ADP-ribose
- IC50
the inhibitor concentration at which an enzymatic reaction velocity is reduced by 50%
- SPPS
solid phase peptide synthesis
- RP-HPLC
reversed-phase high pressure liquid chromatography
- HRMS
high-resolution mass spectrometry
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.05.058.
References and notes
- 1.Chen B, Zang W, Wang J, Huang Y, He Y, Yan L, Liu J, Zheng W. Chem Soc Rev. 2015 doi: 10.1039/c4cs00373j. http://dx.doi.org/10.1039/C4CS00373J. [DOI] [PubMed]
- 2.Ringel AE, Roman C, Wolberger C. Protein Sci. 2014;23:1686. doi: 10.1002/pro.2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. Nat Rev Mol Cell Biol. 2014;15:536. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 4.Greiss S, Gartner A. Mol Cells. 2009;28:407. doi: 10.1007/s10059-009-0169-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin H, Su X, He B. ACS Chem Biol. 2012;7:947. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner GR, Hirschey MD. Mol Cell. 2014;54:5. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seto E, Yoshida M. Cold Spring Harb Perspect Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madsen AS, Olsen CA. Angew Chem Int Ed. 2012;51:9083. doi: 10.1002/anie.201203754. [DOI] [PubMed] [Google Scholar]
- 9.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 10.Sauve AA. Biochim Biophys Acta. 2010;1804:1591. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roth M, Chen WY. Oncogene. 2014;33:1609. doi: 10.1038/onc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HC, Guarente L. Trends Endocrinol Metab. 2014;25:138. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT, Tschöp MH. Physiol Rev. 2012;92:1479. doi: 10.1152/physrev.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osborne B, Cooney GJ, Turner N. Biochim Biophys Acta. 2014;1840:1295. doi: 10.1016/j.bbagen.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Min SW, Sohn PD, Cho SH, Swanson RA, Gan L. Front Aging Neurosci. 2013;5:53. doi: 10.3389/fnagi.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balcerczyk A, Pirola L. BioFactors. 2010;36:383. doi: 10.1002/biof.112. [DOI] [PubMed] [Google Scholar]
- 17.Schemies J, Uciechowska U, Sippl W, Jung M. Med Res Rev. 2010;30:861. doi: 10.1002/med.20178. [DOI] [PubMed] [Google Scholar]
- 18.Zheng W. Mini-Rev Med Chem. 2013;13:132. [PubMed] [Google Scholar]
- 19.Chen L. Curr Med Chem. 1936;2011:18. doi: 10.2174/092986711795590057. [DOI] [PubMed] [Google Scholar]
- 20.Dittenhafer-Reed KE, Feldman JL, Denu JM. ChemBioChem. 2011;12:281. doi: 10.1002/cbic.201000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cen Y. Biochim Biophys Acta. 2010;1804:1635. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch BM, Zheng W. Mol BioSyst. 2011;7:16. doi: 10.1039/c0mb00033g. [DOI] [PubMed] [Google Scholar]
- 23.He B, Hu J, Zhang X, Lin H. Org Biomol Chem. 2014;12:7498. doi: 10.1039/c4ob00860j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fatkins DG, Monnot AD, Zheng W. Bioorg Med Chem Lett. 2006;16:3651. doi: 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 25.Smith BC, Denu JM. Biochemistry. 2007;46:14478. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- 26.Neal RA, Halpert J. Annu Rev Pharmacol Toxicol. 1982;22:321. doi: 10.1146/annurev.pa.22.040182.001541. [DOI] [PubMed] [Google Scholar]
- 27.Ruse MJ, Waring RH. Toxicol Lett. 1991;58:37. doi: 10.1016/0378-4274(91)90188-c. [DOI] [PubMed] [Google Scholar]
- 28.Cox DN, Davidson VP, Judd CE, Stodgell C, Traiger GJ. Toxicol Appl Pharmacol. 1992;113:246. doi: 10.1016/0041-008x(92)90121-8. [DOI] [PubMed] [Google Scholar]
- 29.Coppola GM, Anjaria H, Damon RE. Bioorg Med Chem Lett. 1996;6:139. [Google Scholar]
- 30.Kang JS, Wanibuchi H, Morimura K, Wongpoomchai R, Chusiri Y, Gonzalez FJ, Fukushima S. Toxicol Appl Pharmacol. 2008;228:295. doi: 10.1016/j.taap.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch BM, Hao Y, Li X, Wesdemiotis C, Wang Z, Zheng W. Bioorg Med Chem Lett. 2011;21:4753. doi: 10.1016/j.bmcl.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldman JL, Baeza J, Denu JM. J Biol Chem. 2013;288:31350. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirsch BM, Gallo CA, Du Z, Wang Z, Zheng W. MedChemComm. 2010;1:233. [Google Scholar]
- 34.Takahashi H, Nishina A, Fukumoto RH, Kimura H, Koketsu M, Ishihara H. Life Sci. 2005;76:2185. doi: 10.1016/j.lfs.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 35.Roche Applied Science. Pronase: Product Description. 2006. And references cited therein.
- 36.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Science. 2011;334:806. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, Chandel NS. Proc Natl Acad Sci U S A. 2010;107:8788. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J, Skorski T. Blood. 2006;108:319. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.