

Abstract
Adeno-associated virus (AAV) nonstructural proteins Rep78 and Rep68 carry out all DNA transactions that regulate the AAV life cycle. They share two multifunctional domains: an N-terminal origin binding/nicking domain (OBD) from the HUH superfamily and a SF3 helicase domain. A short linker of ~20 amino acids that is critical for oligomerization and function connects the two domains. Although X-ray structures of the AAV5 OBD and AAV2 helicase domains have been determined, information about the full-length protein and linker conformation is not known. This article presents the solution structure of AAV2 Rep68 using small-angle X-ray scattering (SAXS). We first determined the X-ray structures of the minimal AAV2 Rep68 OBD and of the OBD with the linker region. These X-ray structures reveal novel features that include a long C-terminal α-helix that protrudes from the core of the protein at a 45° angle and a partially structured linker. SAXS studies corroborate that the linker is not extended, and we show that a proline residue in the linker is critical for Rep68 oligomerization and function. SAXS-based rigid-body modeling of Rep68 confirms these observations, showing a compact arrangement of the two domains in which they acquire a conformation that positions key residues in all domains on one face of the protein, poised to interact with DNA.
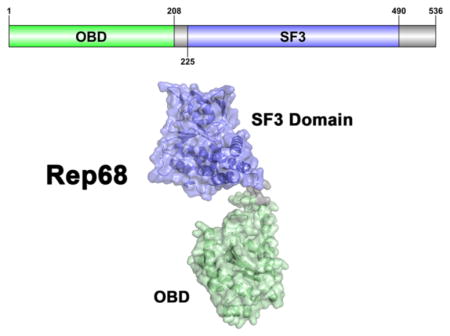
The nonstructural Rep proteins from adeno-associated virus (AAV) are multifunctional proteins with specialized domains equipped to handle the complex interactions with DNA during the AAV life cycle.1,2 AAV has a single-stranded DNA genome of ~4.7 kb containing two major open reading frames (ORFs) flanked by inverted terminal repeats (ITRs). The ITRs form T-shaped hairpin structures and contain the cis-regulatory functions required for replication, transcriptional regulation, and possibly site-specific integration.3–9 The stem of the hairpin forms a double-stranded region containing a Rep binding site (RBS) made up of several 5′-GCTC-3′ repeats.10 A terminal resolution site (trs) located upstream of the RBS is the site of a strand- and site-specific endonuclease reaction required to complete the replication of the AAV genome.3,11,12 Despite its limited genome size, AAV generates eight different polypeptide chains. The right ORF regulated by the P40 promoter produces three capsid proteins (VP1–3) and an assembly activating protein (AAP).13–18 The left ORF uses two different promoters, and alternative splicing, to generate four nonstructural proteins: two large Rep proteins (Rep78 and Rep68) transcribed from the p5 promoter and two small Rep proteins (Rep52 and Rep40) regulated through the p19 promoter.14,19–21 Thus, Rep40 is equivalent to the helicase domain of Rep68. Most of the biochemical activities required for transcriptional regulation, 22,23 DNA replication24,25 and site-specific integration26–28 are carried out by the large Rep78/Rep68 proteins, whereas the small Reps are thought to be important for DNA packaging into preformed capsids.29,30
The wide spectrum of functions performed by Rep78/Rep68 is a direct reflection of the multifunctionality of their domains. The N-terminal origin binding domain (OBD) displays site-and strand-specific endonuclease activity and recognizes the GCTC repeats.24,31–34 Structurally, the OBD is a member of the HUH endonuclease family specialized in the cleavage and rejoining of single-stranded DNA substrates (ssDNA), which occurs during transposition, bacterial conjugation, rolling-circle replication of bacterial DNA and bacteriophages, and in the replication of small eukaryotic viruses.35 This family is characterized by two signature motifs that participate in the nuclease reaction: the HUH motif, which consists of two histidine (H) residues separated by a large hydrophobic residue (U), and the Y motif, with either one or two tyrosine (Y) residues. Structurally, the common feature of HUH endonucleases is a five-stranded antiparallel β-sheet surrounded by α-helices, where the HUH motif is found in one of the strands. The position of the Y motif can vary: in the relaxase subgroup, it is located N-terminally, whereas it is found at the C-terminus in the Rep subclass.36–38
The helicase domain found in all Rep proteins is a representative of the SF3 family helicase with 3′ to 5′ unwinding activity.38–43 The ATPase core is a modified version of the AAA+ domain with additional features that include an N-terminal helical bundle and a β-hairpin that is involved in DNA binding during translocation.38 Consequently, the large Rep proteins can interact with DNA in two different modes: one is mediated by the OBD and recognizes DNA in a sequence-specific manner, whereas the SF3 helicase domain interacts with DNA nonspecifically.44 Furthermore, the large Rep proteins show a complex and dynamic oligomerization behavior, which may add an additional level of regulation to Rep interactions with DNA.45 How the two domains cooperate with each other in order to interact with DNA and how they are arranged in the context of Rep78/Rep68 is not known. Moreover, studies have established that the linker region plays a critical role in the oligomerization and function of AAV Rep proteins; therefore, it is likely that this region acquires a particular secondary structure and may not simply be extended.46,47 To answer these questions, we performed X-ray crystallography and small angle X-ray scattering (SAXS) studies on the AAV2 Rep68 functional domains and full-length protein. Our results show that the OBD has a long extended helix that includes part of the linker, which is not extended, but is tilted in a way that brings the two domains into a compact configuration. SAXS studies on a monomeric version of full-length Rep68 confirm this observation and show for the first time the overall domain architecture of a full-length AAV Rep protein.
MATERIALS AND METHODS
Protein Expression and Purification
The DNA region encoding amino acids 1–208 (OBD) and 1–224 (OBD plus linker) from adeno-associated virus type 2 (AAV2) (GeneBank protein_id = AF043303.1) was cloned into pET15b (Novagen) using restriction sites NdeI and XhoI. The residue C151 was mutated to serine, as it was found to produce disulfide bonds and inhibit crystallization, and Rep-C151S was shown to be fully functional in supporting the AAV life cycle.45 The OBD constructs were overexpressed in Escherichia coli strain BL21 pLysS at 37 °C in Luria–Bertani (LB) broth. IPTG (isopropyl-β-D-thiogalactopyranoside) was added to a final concentration of 1 mM when an OD of 0.6 was reached. Cells were harvested after 5 h and stored at −80 °C. The cell pellets were resuspended in binding buffer (20 mM Tris-HCl, 500 mM NaCl, 10 mM imidazole, 10% glycerol, 1 mM TCEP, pH 7.9) and lysed by sonication. The OBD was purified with a Ni-NTA column (Qiagen) using step gradients of 10 and 30 mM imidazole to wash nonspecific proteins binding to the column and then eluted with 100 mM imidazole. Protein was loaded onto a HiLoad desalting column (GE) to change into thrombin buffer (25 mM Tris-HCl, 200 mM NaCl, 10% glycerol pH 8.0). His-tag was cut by addition of thrombin (1 unit/mg) and removed by passing through a Ni-NTA column. The untagged OBD was collected from the flow through, concentrated, and further purified by gel filtration on a HiLoad 16/60 Superdex 75 column (GE Healthcare) previously equilibrated with gel filtration (GF) buffer (25 mM Tris-HCl, 200 mM NaCl, 1 mM TCEP, pH 7.5). The protein was concentrated to ~40 mg/mL using Millipore Centricon (10 kDa cutoff). AAV2 Rep40 was purified as described elsewhere.38 AAV-2 Rep68wt (1–536) and Rep68Y224Δ were expressed in E. coli strain BL21 pLysS at 18 °C as described in earlier reports.46 In brief, histidine-tagged Rep68 was purified in a Ni-NTA column, and after PreScission protease cleavage of the His-tag, the protein was purified on a HiLoad Superdex 200 16/60 column (GE Healthcare). OBDL was purified using the same procedure.
Crystallization, X-ray Data Collection, and Structure Determination
Crystallization was carried out using the hanging-drop method with commercially available screening kits at 4 °C. Crystals grew after 2 to 3 days in 50 mM cacodylate, pH 6.5, 80 mM sodium acetate, 15 mM magnesium acetate, and 8–10% isopropanol. Crystals were cryoprotected in reservoir buffer and supplemented with 20% MPD before flash freezing them in liquid nitrogen. The crystals diffracted to 2.3 Å and belonged to space group P21212 with unit cell dimensions a = 186.4 Å, b = 154.4 Å, c = 38.8 Å. Diffraction data was collected at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory beamline X6a. The data were processed with the program HKL2000,48 and the structure was solved by molecular replacement using the program PHENIX. We used the structure of the AAV5 OBD as a search model (PDB ID: 1M55). Model building was carried out using PHENIX,49 and manual building, using the program COOT.50 OBDL data was collected using our X-ray home source that consists of a Rigaku Micromax 007 X-ray generator and a Raxis IV+2 area detector.
Sedimentation Velocity
Sedimentation velocity experiments were carried out using a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter Inc.) equipped with an eight-position AN-60Ti rotor. Rep protein samples were loaded in the cells, using, in all cases, the GF buffer. Samples in double sector cells were centrifuged at 25 000 rpm. In all experiments, temperature was kept at 20 °C. Sedimentation profiles were recorded using UV absorption (280 nm) and interference scanning optics. For analysis of the data, the program Sedfit was used to calculate sedimentation coefficient distribution profiles using the Lamm equation.51
Small-Angle X-ray Scattering (SAXS)
Data were collected at three different concentrations that produce a single homogeneous population, as determined by sedimentation velocity studies. Synchrotron SAXS measurements were performed at Brookhaven National Laboratories at beamline X9 and at the Advanced Light Source at the Lawrence Berkeley National Laboratory at the SYBILS beamline. Three different concentrations of each sample were prepared and measured (1, 2, and 3 mg/mL). All data were processed with the package ATSAS.52 Buffer subtraction was carried out using beamline-specific software. Radii of gyration (Rg) were evaluated using the Guinier approximation, sRg < 1.3. Distance distribution functions and maximum diameters Dmax were calculated using the program GNOM.53 SAXS molecular envelopes were calculated using the programs DAMMIN and GASBOR.54,55 Conformational flexibility of the linker and C-terminal tail was analyzed with the program EOM.56 We used the structures of Rep40 (PDB ID: 1s9h) and of OBDL and connect the two domains with a flexible linker (residues 215–224) to generate an atomic model of AAV2 Rep68. Using this initial model, we performed rigidbody and molecular dynamics using BILBOMD to generate the best Rep68 model that fits the SAXS data.57
DNA Helicase Assay
The helicase assay was based on a modification of the strand-displacement assay described elsewhere.58 The DNA substrate (28:18) consists of 3′ tail of 10 nucleotides adjacent to 18 bp. The top strand has been labeled with fluorescein (F) at the 5′ end. All reactions were performed in a final volume of 50 μL in a buffer containing 25 mM HEPES, 50 mM NaCl, pH. 7.0. For the reaction, 1 μM protein was mixed with 0.5 μM ds F-DNA (28:18). The reaction was started by addition of 5 mM ATP-MgCl2 and 2.5 μM trap ssDNA. Reaction was incubated at 25 °C for 1 h. EDTA was used to stop the reaction at a final concentration of 20 mM. Aliquots of 10 μL were loaded in a 12% bis-acrylamide gel (30%) (19:1) using 6× loading dye (0.25 xylene cyanol FF, 30% glycerol). For densitometry and analysis of the bands, a Gel Doc EZ Imager was used, using the automatic lane and band detection tool. Lane background subtraction, white illumination, and an activation time of 300 s were used for the analysis.
Fluorescent Anisotropy Binding Assays
Binding assays were performed using 5 nM fluorescein labeled 41-mer DNA containing the Rep binding site (TGGCGGCGGTTGGGGCTCGGCGCTCGCTCGCTCGCTGGGCG). Rep68 at different concentrations was mixed with DNA at a final volume of 300 μL using the following buffer: 25 mM HEPES (pH 7.0), 200 mM NaCl, 1 mM TCEP. Fluorescence readings were taken on a PC1 fluorimeter (ISS, Inc.) with excitation and emission filters at 492 and 528 respectively. Tubes were equilibrated at 20 °C for 20 min before measurement. Each anisotropy point is the average of 10 measurements. Anisotropy is calculated as the ratio of the difference between vertical and horizontal emission intensities over the total normalized intensity. The fraction of DNA bound (B) was calculated using eq 1
| (1) |
where [A]x represents the anisotropy measured at protein concentration x, [A]DNA is the anisotropy of free fluorescence DNA, and [A]FINAL is the anisotropy at saturation. Data was fit to a single binding site model using the program PRISM6 (GraphPad). Each experiment was done in triplicate.
AAV Infectious Particles Assay
293T cells were transfected with three plasmids using polyethylenimine (PEI): an AAV2 ITR-containing plasmid encoding a CAG-controlled GFP gene (pTRUF11), a helper plasmid expressing AAV2 Rep (wt or L193A-V196A cloned from the pHisRep68LV/15b) and Cap derived from pDG, and a third construct containing the adenovirus helper functions (HGTI plasmid).59,60 The presence of the L193A–V196A double mutation was confirmed by sequencing (Eurofins). After 72 h, the cell supernatant was harvested and increasing volumes of supernatant were used to infect HeLa cells. The percentage of GFP-positive HeLa cells was determined 48 h postinfection by FACS (FACSCanto, BD Biosciences).
RESULTS
We opted to structurally characterize full-length AAV2 Rep68 using small-angle X-ray scattering and rigid-body modeling. A diagram of the different domains and protein constructs used in this study is shown in Figure 1A. We first determine the crystal structure of its OBD including the region spanning the linker, followed by SAXS studies of Rep40 (SF3 helicase domain) to study the dynamic behavior of the C-terminal tail. Finally, to determine the relative orientation of its two functional domains, we determined the SAXS solution structure of Rep68.
Figure 1.

Structure of AAV2 OBD. (A) Domain structure of AAV2 Rep68 protein: OBD is shown in green, Rep40 (SF3 helicase domain), in blue, and linker and C-terminal tail, in gray. The Rep40 protein spans residues 225–536 of Rep68. (B) Topology diagram of AAV2 OBD. (C) Ribbon diagram of the OBD structure. The α-helices are light green, and β-strands are dark green. Secondary structure elements are labeled with α-helices A–F, and β-sheets are numbered 1–5. The DNA binding loop LDB with missing residues in the structure is shown as a dotted line. (D) A view of the structure rotated by 90° clockwise. α-Helix F is shown protruding from the core of the structure at an angle of almost 45°.
Structure of AAV-2 Rep OBD
The OBD construct (residues 1–208) was initially obtained from limited proteolysis experiments.61 The structure was solved by molecular replacement and refined to 2.3 Å resolution (Table 1). There are three molecules in the asymmetric unit: molecule A, spanning residues 1–138 and 143–206, molecule B, spanning residues 1–139 and 143–201, and molecule C, covering residues 1–131 and 141–191. The missing residues are all part of the DNA binding loop (LDB). Molecules A and B form a dimer interacting through their C-terminal helix F, whereas molecule C does not make any close contacts. The OBD structure is characterized by a central five-stranded antiparallel β-sheet flanked by six α-helices, three at each side of the sheet (Figure 1B). The topology of the β-sheet follows a 4–1–3–2–5 order, with helices E and F on one side of the sheet and helices B–D positioned on the opposite side (Figure 1C,D). There are two long loops: the DNA binding loop LDB connects β-strand 4 to α-helix E and loop LBC connects α-helices B and C and protrudes underneath α-helix F (Figure 1C). LDB was identified as being important in making specific contacts with the major groove of GCTC repeats and shows the highest degree of variation among all AAV Rep isoforms.31,62 This is a dynamic region, as shown by the lack of electron density in all three molecules found in the asymmetric unit. The C-terminal helix αF is long, spanning residues 182–206, and protrudes at an approximately 45° angle from the plane of the β-sheet (Figure 1D). The residues at the end of helix F appear to be dynamic, as we could see only up to residue 201 in molecule B and to residue 191 in molecule C. Helix F is loosely packed against helix E and the loop connecting β5 to αF. The interactions that keep these three elements together are sparse, suggesting that helix F is flexible. Several dynamic regions in the AAV2 OBD can be observed by the superposition of the three molecules in the asymmetric unit (Figure 2A). The three molecules superimpose with an overall rmsd over 197 Cα of 0.62 Å. Two regions can be identified with larger than average values: region 1 includes resides 15–34 spanning helices A and B, and region 2 includes residues from LDB. Not surprisingly, based on the AAV5 OBD–RBS structure, both of these regions are involved in DNA interactions: region 1 is involved in recognition of the ITR hairpin stem 2, and LDB recognizes part of the GCTC repeat.31 Thus, folding and stabilization of these elements must occur upon DNA binding. Two of the OBD molecules in the asymmetric unit form a dimer through interactions with residues present in α-helix F, resembling a pseudocoiled coil (Figure 2B). These include L193, V196, H192, and S197 making hydrophobic and hydrogen-bond interactions (Figure 2C). This type of interface was also observed in the AAV5 OBD–RBS complex, which may suggest a functional role for this interface in the context of full-length Rep78/Rep68 proteins.31 To determine if these interactions are functionally relevant in Rep68 function and their effect on the AAV life cycle, we examined the ability of several Rep68 mutants to oligomerize, to unwind dsDNA, and to support the production of infectious particles. Figure 2D shows that Rep68 mutants H192A, S197A, and L193AV196A have similar sedimentation velocity profiles as that of Rep68wt, forming the 13S peak that corresponds to heptameric/octameric ring species.45 In addition, the mutants have similar unwinding activity as that of the wild-type protein (Figure 2E). Thus, the different mutants are functionally equivalent to Rep68wt. This conclusion is supported by the ability of the Rep68-L193AV196A double mutant to produce infectious viral particles as efficiently as wild-type Rep68 (Figure 2F). These results show that the dimer interactions are generated during crystal packing and are not involved in Rep68 oligomerization and function.
Table 1.
Data Collection and Refinement Statistics
| OBD (1–208) | OBDL (1–224) | |
|---|---|---|
| space group | P21212 | P21 |
| Cell Dimensions | ||
| a, b, c (Å) | 186.4, 154.4, 38.8 | 75.6, 178.7, 130.4 |
| α, β, γ (deg) | 90, 90, 90 | 90, 91.7, 90 |
| wavelength (Å) | 0.9792 | 1.54 |
| resolution (Å) | 30–2.30 (2.34–2.30) | 30–2.6 (2.69–2.6) |
| no. of measured | 566 473 | 426 030 |
| no. of unique | 50 960 | 105 074 |
| data coverage (%)a | 99.8 (100) | 99.1 (98.9) |
| Rmerge(%)a,b | 0.099 (0.402) | 0.075 (0.311) |
| I/σa | 17.7 (8.6) | 12.4 (4.1) |
| refinement statistics | ||
|---|---|---|
| resolution range | 30.0–2.3 | 29.8–2.6 |
| reflections | 50 960 | 105 007 |
| Rwork (%)c | 22.5 | 19.1 |
| Rfree (%)d | 24.2 | 23.8 |
| non-hydrogen atoms | 5024 | 17677 |
| protein | 4704 | 17493 |
| metal | 2 | 11 |
| water | 314 | 173 |
| average B-factors (Å2) | 49.8 | 59.1 |
| protein | 50.0 | 59.2 |
| metal | 49.2 | 88.4 |
| water | 46.4 | 47.3 |
| RMS Deviations | ||
| bonds (Å) | 0.02 | 0.003 |
| angles (Å) | 1.4 | 1.53 |
| Ramachandran Plot Quality | ||
| most favored (%) | 99.0 | 97.0 |
| additional allowed (%) | 1.0 | 3.0 |
| generously allowed (%) | 0 | 0.0 |
| disallowed (%) | 0 | 0 |
Values for the outermost shells are given in parentheses.
Rmerge = Σ|I − 〈I〉|/Σ I, where I is the integrated intensity of a given reflection.
Rwork = Σ[||Fo| − |Fc||Σ|Fo|.
For Rfree calculations, 5% of data was excluded from refinement.
Figure 2.

Superposition of OBD molecules in the asymmetric unit. (A) Ribbon diagram of the superposition of the three molecules in the asymmetric unit. Molecule A is shown in green; molecule B, blue; and molecule C, red. The three loops LDB have different conformations and are shown as dotted connections. (B) Ribbon diagram of the dimer in the asymmetric unit formed by molecules A and B. The small dimer interface occurs through residues in α-helix F. (C) Details of the residues involved in the oligomeric interaction with Ser197, Val196, and Leu193 from molecule A interacting with His192, Leu193, and Val196 from molecule B. (D) Sedimentation velocity profiles of Rep68wt, Rep68-L193AV196A, Rep68-H192A, and Rep68-S197A. Experiments were carried out at 20 °C and 25 000 rpm on a Beckman XL-I analytical ultracentrifuge. Scans were collected every 2 min using absorbance at 280 nm. Data was analyzed using the program Sedfit.51 (E) Helicase assay of Rep68 mutants. Fluorescent-labeled DNA molecule has a 3′ single-stranded tail and an 18 bp region (first lane). Upon ATP addition, helicase activity is shown as the 18 nucleotide fluorescein-labeled ssDNA is displaced (second lane). Rep68, L193AV196A, H192A, and S197 proteins all unwind DNA. K340H mutant is ATPase-negative and does not unwind DNA. Last lane shows the DNA substrate after heating at 100 °C for 5 min. (F) Comparison of the production of rAAV2-GFP infectious particles in the presence of wt or LV mutant. Various volumes of supernatant (in (μ), x axis) from 293T cells producing rAAV2-GFP were added to HeLa cells, and the percentage of GFP-positive infected cells was determined by FACS analysis, as described in Materials and Methods. Data is presented as average ± standard deviation from three independent experiments.
Structural comparison of the AAV2 and AAV5 OBD
The Rep proteins from the majority of AAV serotypes show an overall homology with the AAV2 OBD of 95% (Figure S1). In contrast, AAV5 Rep has only 59% identity over the N-terminal 224 residues. However, the AAV2 and AAV5 OBD structures superimposed well, with an overall rmsd of 0.94 Å for 191 aligned Cα carbons. As shown in Figure 3A,B, there are, nonetheless, three regions that show differences: region A (residues 12–15) includes the linker connecting β1 to αA, region B (74–77) spans the turn connecting β3 to αD, and region C (139–147) consists of LDB. Among all AAV serotypes, AAV5 and AAV8 have the largest sequence variations. For instance, AAV5 has a shorter LDB loop than AAV2, whereas in AAV8, the loop is longer by 2 residues. In addition, the AAV5 protein has four additional residues after position 198. Moreover, sequence conservation in the αF region is low in AAV5, suggesting that this structure may indeed be shorter than in AAV2 (Figure S1). The overall electrostatic surface representations of the two proteins look similar, but subtle differences are visible. In both structures, one side of the surface is highly positively charged; however, in AAV2 OBD, the positive patch is wider and more intense. This is seen more explicitly if we compare the positive surface potential values between the two OBDs, with 2130.6 kcal/molq for AAV2 and 1720.4 kcal/molq for AAV5.63 This area includes LDB, helix D, and the N-terminal half of α-helix C. The former two interact with the GCTC repeats, whereas the latter is involved in the recognition of the RBE’ hairpin in the ITRs.31 Helix F is mostly negatively charged, but in AAV2, there is a slightly positive patch located in the first three turns that is not visible in AAV5 (Figure 3C). Because the structure of the AAV5 construct includes up to residue 197 and the sequence homology around this region is less than 80%, it is not apparent whether helix F in AAV5 will be as extended as in AAV2. Whether these structural and sequence variations found in LDB and the linker region account for the differences in nicking specificity observed for AAV2 and AAV5 Rep proteins remains to be determined and will require structures with DNA substrates along the nicking reaction pathway.64,65
Figure 3.

Comparison of AAV2 and AAV5 OBD structures. (A) Ribbon diagram of the superposition of the AAV2 (green) and AAV5 (blue) OBD structures. The N and C termini are marked. Regions that have the largest rmsd are indicated by arrows. (B) Plot of rmsd differences against residue number between AAV2 and AAV5 OBD. (C) Solvent-accessible electrostatic surface comparison of AAV2 and AAV5 OBDs showing three views of 90° counterclockwise rotations. Positive regions are shown in blue, and negative, in red.
Crystal Structure of OBD-Linker
We and others have described previously that the linker region connecting the OBD and the SF3 helicase domain of the large Rep proteins is pivotal for the assembly of multimeric Rep protein complexes.46,47 To better characterize this region, we expressed, purified, and crystallized a construct containing the OBD and the linker that we termed OBDL, spanning residues 1–224. OBDL crystals diffracted to 2.6 Å and belong to space group P21, with 11 molecules in the asymmetric unit. The electron density corresponding to the C-terminus of helix F and the linker region varies among the different molecules in the asymmetric unit; nevertheless, we were able to build a model of OBDL up to residue 214 (Figure 4). We did not detect any electron density from residues 215–224. The structure shows that helix F spans to residue 205 and is followed by a loop that makes a downward turn toward the core of the protein at proline 209 (Figure 4B). Superposition of OBD with OBDL shows that helix F is flexible and moves as a rigid body with respect to the core of the protein, pivoting around the loop that connects it with strand β5. While the two OBD structures superimpose with an overall rmsd of ~0.8 Å, the two αF helices superimpose with an rmsd of only 1.3 Å and appear at a different angle with respect to the core of the protein, with a difference between them of ~2.4° (Figure 4C). The OBDL structure also implies that the linker is not in an extended conformation but, instead, has a particular structure that is constrained by the two prolines in the linker. This particular conformation may bring the two domains together in the context of full-length Rep68. To test the importance of the proline residues, we mutated proline 214 to alanine and measured the effect of this on Rep68’s properties. Figure 4D shows that this mutation drastically alters the oligomeric behavior of Rep68 and affects its ability to form the 13S oligomer species (Figure 4D). Moreover, the mutation affects the ability of Rep68 to bind double-stranded DNA containing a Rep binding site (RBS). Figure 4E shows that the Rep68-P214A mutant binds DNA ~ 7 times less than wild-type protein. Taken together, our results show that the conformation of the linker is critical for the function of Rep68.
Figure 4.

Structure of OBDL. (A) Ribbon diagram of the structure spanning residues 1–214. (B) Electron density of region 206–214 contoured at 0.9σ. The density fits most of the residues, but it suggests that the region is dynamic. (C) Superposition of OBD and OBDL structures illustrating the flexibility of helix F. (D) Comparison of the sedimentation velocity profiles between Rep68wt (top) and Rep68-P214A (bottom). (E) Comparison of binding isotherms of Rep68wt and Rep68-P214A. Fluorescent anisotropy binding assays were carried out with a 41-mer AAVS1 DNA labeled with fluorescein. Apparent dissociation constants were determined from triplicate experiments and fitted to a one binding site-specific binding model.
SAXS Studies of OBDL Validate the Conformation of the Linker
To gain further structural information about the linker domain and to validate the OBDL X-ray structure, we performed solution studies on OBD and OBDL. The concentration of NaCl in the buffer was kept at 0.5–1 M to prevent formation of oligomers induced by the presence of the linker.46 The calculated sedimentation coefficient for both constructs is ~2.0 S (Figure S2). The small difference in S value suggests that the linker region is not in an extended conformation. However, the smaller sedimentation velocity coefficient of OBDL implies a slightly more elongated shape. We subjected both constructs to SAXS studies under conditions that generated single species, as determined by sedimentation velocity. Scattering profiles from three different concentrations were collected, and a final scattering curve was obtained by merging the best curves using the program Almerge.66 The scattering profiles for OBD and OBDL are shown in Figure 5A. The merged data was used to calculate both the radius of gyration (Rg) and the P(r) distribution function as described in the Material and Methods (Figure 5B). Ab initio models were produced using the programs DAMMIN and GASBOR. For each protein, 10 independent models were generated and averaged with Damaver. The final models from the two independent reconstructions were aligned using the program Supcomb, resulting in an overall normalized spatial discrepancy (NSD) value of 0.54, suggesting an excellent correlation between the two independent reconstructions.67 The resulting GASBOR models for OBD and OBDL are shown in Figure 5C with the superimposed X-ray structures of the corresponding constructs. The calculated Rg and Dmax values are similar for both models (Table 2). The OBDL GASBOR ab initio model shows an envelope that resembles a round cylinder, suggesting that, under our experimental conditions, the region between 208 and 224 is not extended but may resemble the overall conformation seen in the OBDL crystal structure (Figure 5C, right panel). To further corroborate the hypothesis that the linker is neither extended nor highly flexible, we carried out molecular dynamics using BILBOMD to determine if an ensemble with multiple conformations of the linker described the SAXS scattering profile of OBDL.57 The results show that inclusion of more than one conformation does not improve the fit to the experimental data significantly (data not shown).
Figure 5.
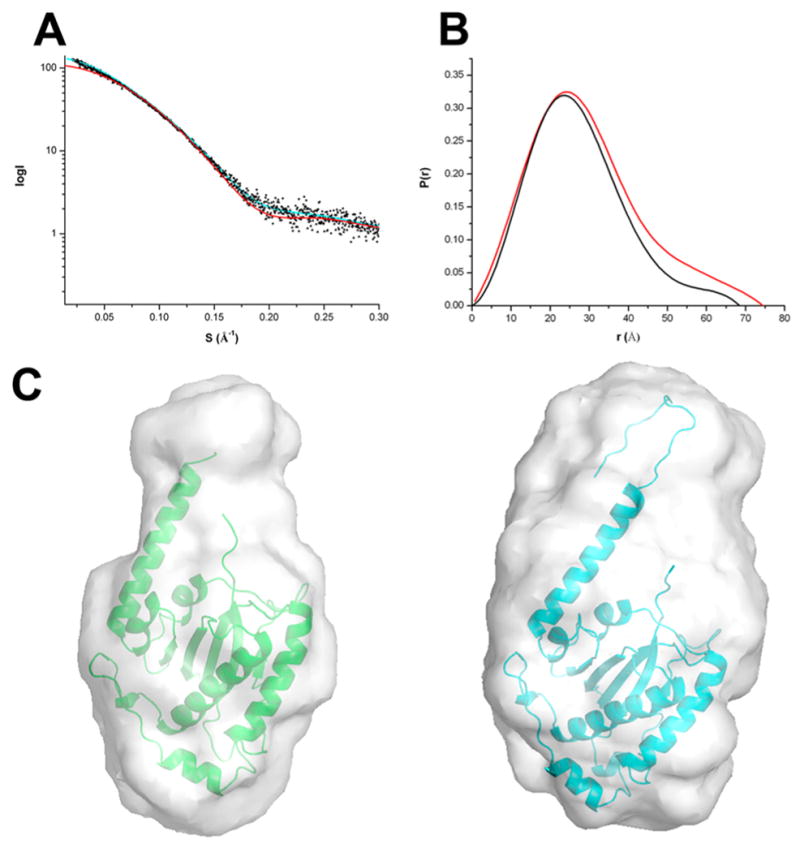
SAXS analysis for OBD and OBDL. (A) SAXS scattering data from OBD (blue) and OBDL (red) in solution. Superimposed is the scattering calculated from the atomic models for OBD (■) and OBDL (△). (B) Normalized pair distribution functions P(r) for OBD (black) and OBDL (red) calculated from the experimental scattering curves using the program PRIMUS. (C) Low-resolution envelopes of DAMMIN and GASBOR models for OBD (left) and OBDL (right) are superimposed with the X-ray structure of OBD and OBDL, respectively.
Table 2.
Hydrodynamic Parameters
| parameter | OBD (1–208) | OBDL (1–224) | Rep40wt (225–536) | Rep68Y224AΔ (1–490) |
|---|---|---|---|---|
| S (s)a | 2.1 | 1.97 | ndd | nd |
| Rg (nm)b | 21.5 ± 0.3 | 23.2 ± 2.3 | 27.6 ± 3.8 | 38.9 ± 0.1 |
| Dmax (nm)c | 68.2 | 74.4 (104.96) | 98.8 | 123.8 |
| χ | 1.0e | 1.9e | 0.5f | 0.8g |
Determined from sedimentation velocity data.
Determined from Guinier analysis.
Determined from P(r) analysis.
nd, not determined.
Experimental/model data fit using FoXS.74
Experimental/model data fit using EOM.
Experimental/model data fit using CORAL.
Modeling Flexibility of C-Terminal Tail of Rep68
AAV Rep proteins Rep40 and Rep68 have a C-terminal tail of 46 residues that extends from the core of the helicase domain, as determined by limited proteolysis experiments.38 To determine the overall flexibility of the C-terminal tail, we carried out SAXS experiments of Rep40wt at different concentrations, and data were analyzed as described previously. A GASBOR model of Rep40 clearly resembles the overall shape of the helicase domain and has a characteristic flat disk shape with an additional elongated density that is not represented in the X-ray structure, which should correspond to the C-terminal tail (Figure 6A). However, the dimensions are smaller than those expected from a fully extended C-terminal tail. To analyze the flexibility of the tail, we used EOM software to search for an optimal ensemble of conformations that will best fit the scattering data. This method generates a large pool of models (10 000) with random conformations and uses a genetic algorithm to select an optimized ensemble of configurations that best describes the SAXS scattering data (Figure 6B).56,68 The best ensemble consisted of four models that fit the experimental curve with a χ of 0.5 (Figure 6C). However, the narrowness of the ensemble Rg distribution suggests that a limited number of conformations are preferred, with one configuration in particular accounting for about 50% of the population. Interestingly, all of the selected models have the C-terminal oriented toward the opposite site of the presensor 1 β-hairpin (PS1βH), a motif that is involved in DNA interactions during DNA translocation and unwinding (Figure 6D).30,69 Thus, our study shows that the C-terminal tail, although flexible, has a preferred set of dominant conformations.
Figure 6.

SAXS analysis of Rep40/Rep68 C-terminal tail. (A) Shape reconstruction of Rep40wt and the docked X-ray structure of Rep40. (B) Comparison of the Rg distribution of the initial EOM generated pool (black line) and the selected model ensemble (red line). (C) Scattering curve of Rep40wt (black) and fit of the optimal EOM model ensemble (red). (D) Superposition of the ensemble generated model showing the preferred conformations of the C-terminal tails.
SAXS-Based Structural Modeling of Rep68 Shows a Compact Conformation in Solution
We previously showed that Rep68 has complex dynamic oligomeric behavior in solution and is present as a mixture of multiple oligomers, including heptameric and octameric rings.45 To obtain a homogeneous population of Rep68 monomers, we took advantage of the Y224A mutation that affects its tendency to form multiple oligomeric species.46 The Rep68Y224A protein is present as a monomer at concentrations less than 5 mg/mL and under high salt conditions. Moreover, we have determined that this mutation has helicase activity comparable to that of the wild-type protein and does not cause any significant structural change.70 To facilitate model building and data interpretation, we used a truncated Rep68 (1–490) to eliminate the C-terminal tail residues.38 SAXS data was collected at 2, 3, and 4 mg/mL and processed as described in the previous sections (Figure 7A). The GASBOR generated model produces an elongated prolate ellipsoid that is slightly curved with dimensions 140 × 73 × 37 Å3 (Figure 7B). Individual atomic structures of OBDL and Rep40 can be easily docked into the envelope; in particular, one end of the particle has the shape of a flat disk that resembles the GASBOR Rep40 ab initio model (Figure 7C). We performed rigid-body modeling using the X-ray structures of OBDL (1–214) and Rep40 (225–490) with CORAL.71 The domain boundaries define an unstructured linker spanning residues 215–224. However, secondary structure prediction suggests that the region between 220 and 224 could form a α-helix extending into the first helix of the helicase domain.46 Consequently, we generated multiple models that extended this helix at different positions. The Rep68 models fit the experimental data very well, with χ values in the range of 1.1–0.8. All models show that the two domains are positioned such that the long axis of the helicase domain is almost perpendicular to the OBD, as shown in Figure 7E. The best model was obtained by extending the N-terminal helicase helix to residue 223. Two main observations can be drawn from this model. First, the orientation of the two domains results in a Rep68 structure where the motifs that interact with DNA in both the OBD (LDB and helix C) and helicase domain (β-hairpin 1) are on the same face of the protein. Second, the two domains are closely positioned, making an extended linker structure unlikely. To further support this conclusion, we assessed interdomain flexibility of Rep68 using two methodologies. First, a Porod–Debye plot of the scattering data shows a plateau that is a signature for a compact molecule.72 The data fits the linear region with a Porod coefficient of 4, again consistent with a compact molecule (Figure 7D). In addition, we carried out BILBOMD and determined that a single model fits the data equally as well as with multiple conformations (Figure 7A).
Figure 7.

SAXS modeling of Rep68. (A) Fit of the experimental SAXS data (black circles) with the theoretical scattering profile obtained from CORAL model (red line) with a χ value of 0.8. (B) Two views of the GASBOR averaged molecular envelope for Rep68Δ showing the approximate dimensions in angstroms. (C) Docking of OBDL and Rep40 atomic structures to the GASBOR ab initio envelope. (D) Porod–Debye plot of Rep68Y224AΔ SAXS data (black squares) supporting a compact protein with little flexibility. Red line represents the linear plateau. (E) Three views of the CORAL rigid-body final model of Rep68Y224AΔ.
DISCUSSION
Our results show new structural features that increase our knowledge of the architecture of AAV Rep proteins. The combined X-ray and SAXS studies show that the AAV2 Rep68 linker region is partially structured, with helix F of the OBD extending until residue 209 and protruding from the main core at a 45° angle. The presence of two proline residues (209 and 214) seems to impart a certain rigidity to the loop region, making a small turn before continuing toward the helicase domain (Figure 4). This conformation is important for Rep68 function, as mutation of one of the proline residues in the linker (P214) is sufficient to prevent proper oligomerization and DNA binding. In addition, part of the remaining linker may extend the first helix of the helical domain of Rep52/40.46 Both of these structural features bring the OBD and helicase domain closer to each other. However, although the linker is not extended, it allows for a certain degree of flexibility. Our rigidbody modeling generated different models with slightly different conformations of the two domains that fit the scattering data equally well (Figure S3). This suggests a certain degree of conformational flexibility between the two domains. Structurally, this flexibility originates from the linker, the OBD helix F, and the helical bundle of the helicase domain. Alignment of the OBD and OBDL structures shows that helix F can pivot and move relative to the main core of the domain (Figure 4C). Likewise, alignment of the three Rep40 molecules found in the asymmetric unit of the X-ray structure (PDB ID: 1s9h) shows the helical bundle at different positions relative to the AAA+ domain (data not shown). This flexibility is important to accommodate changes occurring upon DNA binding and oligomerization. Moreover, analysis of the flexibility of the C-terminal tail in Rep68 suggest that there is a preference in the conformations acquired by the C-terminal tail that positions it at the opposite end of the β-hairpin. This is important in the context of formation of Rep68 oligomeric rings because other conformations may produce steric clashes and inhibit their formation.
Finally, our Rep68 model closely resembles the configuration of the helicase and OBD in one of the molecules seen in the X-ray structure of the SV40 large T antigen (SV40-LTag) in complex with DNA.73 This arrangement results in critical residues that interact with DNA being located on one face of the protein (Figure 8B). In our Rep68 model, these regions include loop LDB and helix αD in the OBD, the helicase domain (PS1β residues K404 and K406), and linker residues R217 and K219 that have been shown to play a role in complex formation and DNA binding in AAV5.47 In addition, the SV40-LTag structure shows that the helical bundle (Zn domain in SV40-LTag) interacts with DNA, docking into the major groove.73 Our model shows that the position of the helical bundle is similar, and it is intriguing to postulate that the conformation of Rep68 is prealigned to interact with DNA in way similar to that of SV40-LTag (Figure 8A).
Figure 8.

Rep68 DNA interacting face. (A) Comparison of Rep68 model (left) and SV40-LTag (right). Colored equivalently are the OBD domain (green) and the helicase domain (blue). (B) (left) Rep68 structure showing the regions that interact with DNA colored in pink. Orientation of Rep68 is 90° counterclockwise from that in panel A. (right) Surface representation of Rep68 showing electropositive regions in blue and negative regions in red. View is in the same orientation as that on the left.
In conclusion, we have completed the first structural description of full-length AAV2 Rep68 protein, revealing the orientation and relative position of its functional domains. These observations provide new clues to explain its DNA binding mode; however, our model does not discard possible changes that should occur upon DNA binding and/or oligomerization. These questions require future structural studies of high-resolution structures of Rep68 oligomers alone and in complex with DNA.
Supplementary Material
Acknowledgments
Funding
This work was supported by NIH grant R01-GM092854 (C.R.E.) and UK MRC grant 1001764 (R.M.L.).
We would like to thank members of NSLS beamlines X6a (Vivian Stojanoff, Jean Jakoncic, and Edwin Lazo) and X9 (Lin Yang and Marc Allaire) and ALS SIBYLS beamline (Kevin Dyer and Kathryn Burnett) for help in data collection. We thank Vishaka Santosh for helpful comments on the preparation of this manuscript.
ABBREVIATIONS
- AAV
adeno-associated virus
- OBD
origin binding domain
- OBDL
origin binding domain with linker
- HUH
histidine–hydrophobic–histidine
- DBD
DNA binding domain
- SF3
superfamily 3 helicase
- SAXS
small-angle X-ray scattering
- ORF
open reading frame
- ITR
inverted terminal repeat
- RBS
Rep binding site
- ssDNA
single-stranded DNA
- AAA+
ATPases associated with various cellular activities
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LB
Luria–Bertani
- TCEP
tris(2-carboxyethyl)phosphine
- NTA
nitrilotriacetic acid
- MPD
2-methyl-2,4-pentanediol
- NSLS
National Synchrotron Light Source
- Rg
radius of gyration, PEI, polyethylenimine
- FACS
fluorescent activated cell sorting
Footnotes
Notes
The authors declare no competing financial interest.
Accession Codes
PDB ID Codes: 4ZO0, 5DCX
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00610.
Sequence and structural alignment of the AAV OBD serotypes, sedimentation velocity profiles of AAV2 OBD and OBDL, and superposition of different Rep68 models obtained by independent CORAL runs (PDF).
References
- 1.Henckaerts E, Linden RM. Adeno-associated virus: a key to the human genome? Future Virol. 2010;5:555–574. doi: 10.2217/fvl.10.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weitzman MD, Linden RM. Adeno-associated virus biology. Methods Mol Biol. 2012;807:1–23. doi: 10.1007/978-1-61779-370-7_1. [DOI] [PubMed] [Google Scholar]
- 3.Lusby E, Fife KH, Berns KI. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J Virol. 1980;34:402–409. doi: 10.1128/jvi.34.2.402-409.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samulski RJ, Srivastava A, Berns KI, Muzyczka N. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell. 1983;33:135–143. doi: 10.1016/0092-8674(83)90342-2. [DOI] [PubMed] [Google Scholar]
- 5.Senapathy P, Tratschin JD, Carter BJ. Replication of adeno-associated virus DNA. Complementation of naturally occurring rep- mutants by a wild-type genome or an orimutant and correction of terminal palindrome deletions. J Mol Biol. 1984;179:1–20. doi: 10.1016/0022-2836(84)90303-6. [DOI] [PubMed] [Google Scholar]
- 6.Beaton A, Palumbo P, Berns KI. Expression from the adeno-associated virus p5 and p19 promoters is negatively regulated in trans by the rep protein. J Virol. 1989;63:4450–4454. doi: 10.1128/jvi.63.10.4450-4454.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira DJ, McCarty DM, Muzyczka N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J Virol. 1997;71:1079–1088. doi: 10.1128/jvi.71.2.1079-1088.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surosky RT, Urabe M, Godwin SG, McQuiston SA, Kurtzman GJ, Ozawa K, Natsoulis G. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J Virol. 1997;71:7951–7959. doi: 10.1128/jvi.71.10.7951-7959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young SM, Jr, Samulski RJ. Adeno-associated virus (AAV) site-specific recombination does not require a Repdependent origin of replication within the AAV terminal repeat. Proc Natl Acad Sci U S A. 2001;98:13525–13530. doi: 10.1073/pnas.241508998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarty DM, Ryan JH, Zolotukhin S, Zhou X, Muzyczka N. Interaction of the adeno-associated virus Rep protein with a sequence within the A palindrome of the viral terminal repeat. J Virol. 1994;68:4998–5006. doi: 10.1128/jvi.68.8.4998-5006.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauswirth WW, Berns KI. Origin and termination of adeno-associated virus DNA replication. Virology. 1977;78:488–499. doi: 10.1016/0042-6822(77)90125-8. [DOI] [PubMed] [Google Scholar]
- 12.Spear IS, Fife KH, Hauswirth WW, Jones CJ, Berns KI. Evidence for two nucleotide sequence orientations within the terminal repetition of adeno-associated virus DNA. J Virol. 1977;24:627–634. doi: 10.1128/jvi.24.2.627-634.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becerra SP, Rose JA, Hardy M, Baroudy BM, Anderson CW. Direct mapping of adeno-associated virus capsid proteins B and C: a possible ACG initiation codon. Proc Natl Acad Sci U S A. 1985;82:7919–7923. doi: 10.1073/pnas.82.23.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becerra SP, Koczot F, Fabisch P, Rose JA. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J Virol. 1988;62:2745–2754. doi: 10.1128/jvi.62.8.2745-2754.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cassinotti P, Weitzand M, Tratschin JD. Organization of the adeno-associated virus (AAV) capsid gene: mapping of a minor spliced mRNA coding for virus capsid protein. Virology. 1988;167:176–184. [PubMed] [Google Scholar]
- 16.Trempe JP, Carter BJ. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J Virol. 1988;62:3356–3363. doi: 10.1128/jvi.62.9.3356-3363.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muralidhar S, Becerra SP, Rose JA. Site-directed mutagenesis of adeno-associated virus type 2 structural protein initiation codons: effects on regulation of synthesis and biological activity. J Virol. 1994;68:170–176. doi: 10.1128/jvi.68.1.170-176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonntag F, Schmidt K, Kleinschmidt JA. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A. 2010;107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green MR, Roeder RG. Transcripts of the adeno-associated virus genome: mapping of the major RNAs. J Virol. 1980;36:79–92. doi: 10.1128/jvi.36.1.79-92.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green MR, Straus SE, Roeder RG. Transcripts of the adenovirus-associated virus genome: multiple polyadenylated RNAs including a potential primary transcript. J Virol. 1980;35:560–565. doi: 10.1128/jvi.35.2.560-565.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laughlin CA, Westphal H, Carter BJ. Spliced adenovirus-associated virus RNA. Proc Natl Acad Sci U S A. 1979;76:5567–5571. doi: 10.1073/pnas.76.11.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyostio SR, Wonderling RS, Owens RA. Negative regulation of the adeno-associated virus (AAV) P5 promoter involves both the P5 rep binding site and the consensus ATP-binding motif of the AAV Rep68 protein. J Virol. 1995;69:6787–6796. doi: 10.1128/jvi.69.11.6787-6796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dutheil N, Smith SC, Agundez L, Vincent-Mistiaen ZI, Escalante CR, Linden RM, Henckaerts E. Adeno-associated virus Rep represses the human integration site promoter by two pathways that are similar to those required for the regulation of the viral p5 promoter. J Virol. 2014;88:8227–8241. doi: 10.1128/JVI.00412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Im DS, Muzyczka N. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell. 1990;61:447–457. doi: 10.1016/0092-8674(90)90526-k. [DOI] [PubMed] [Google Scholar]
- 25.Chiorini JA, Weitzman MD, Owens RA, Urcelay E, Safer B, Kotin RM. Biologically active Rep proteins of adeno-associated virus type 2 produced as fusion proteins in Escherichia coli. J Virol. 1994;68:797–804. doi: 10.1128/jvi.68.2.797-804.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weitzman MD, Kyostio SR, Kotin RM, Owens RA. Adeno-associated virus (AAV) Rep proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc Natl Acad Sci U S A. 1994;91:5808–5812. doi: 10.1073/pnas.91.13.5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urcelay E, Ward P, Wiener SM, Safer B, Kotin RM. Asymmetric replication in vitro from a human sequence element is dependent on adeno-associated virus Rep protein. J Virol. 1995;69:2038–2046. doi: 10.1128/jvi.69.4.2038-2046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamartina S, Ciliberto G, Toniatti C. Selective cleavage of AAVS1 substrates by the adeno-associated virus type 2 rep68 protein is dependent on topological and sequence constraints. Journal of virology. 2000;74:8831–8842. doi: 10.1128/jvi.74.19.8831-8842.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King JA, Dubielzig R, Grimm D, Kleinschmidt JA. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 2001;20:3282–3291. doi: 10.1093/emboj/20.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon-Robarts M, Blouin AG, Bleker S, Kleinschmidt JA, Aggarwal AK, Escalante CR, Linden RM. Residues within the B′ motif are critical for DNA binding by the superfamily 3 helicase Rep40 of adeno-associated virus type 2. J Biol Chem. 2004;279:50472–50481. doi: 10.1074/jbc.M403900200. [DOI] [PubMed] [Google Scholar]
- 31.Hickman AB, Ronning DR, Perez ZN, Kotin RM, Dyda F. The nuclease domain of adeno-associated virus rep coordinates replication initiation using two distinct DNA recognition interfaces. Mol Cell. 2004;13:403–414. doi: 10.1016/s1097-2765(04)00023-1. [DOI] [PubMed] [Google Scholar]
- 32.Yoon-Robarts M, Linden RM. Identification of active site residues of the adeno-associated virus type 2 Rep endonuclease. J Biol Chem. 2003;278:4912–4918. doi: 10.1074/jbc.M209750200. [DOI] [PubMed] [Google Scholar]
- 33.Owens RA, Weitzman MD, Kyostio SR, Carter BJ. Identification of a DNA-binding domain in the amino terminus of adeno-associated virus Rep proteins. J Virol. 1993;67:997–1005. doi: 10.1128/jvi.67.2.997-1005.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hickman AB, Ronning DR, Kotin RM, Dyda F. Structural unity among viral origin binding proteins: crystal structure of the nuclease domain of adeno-associated virus Rep. Mol Cell. 2002;10:327–337. doi: 10.1016/s1097-2765(02)00592-0. [DOI] [PubMed] [Google Scholar]
- 35.Koonin EV, Ilyina TV. Computer-assisted dissection of rolling circle DNA replication. BioSystems. 1993;30:241–268. doi: 10.1016/0303-2647(93)90074-m. [DOI] [PubMed] [Google Scholar]
- 36.Chandler M, de la Cruz F, Dyda F, Hickman AB, Moncalian G, Ton-Hoang B. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol. 2013;11:525–538. doi: 10.1038/nrmicro3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Prieto C, Agundez L, Linden RM, Llosa M. HUH site-specific recombinases for targeted modification of the human genome. Trends Biotechnol. 2013;31:305–312. doi: 10.1016/j.tibtech.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 38.James JA, Escalante CR, Yoon-Robarts M, Edwards TA, Linden RM, Aggarwal AK. Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure. 2003;11:1025–1035. doi: 10.1016/s0969-2126(03)00152-7. [DOI] [PubMed] [Google Scholar]
- 39.Gorbalenya AE, Koonin EV, Wolf YI. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990;262:145–148. doi: 10.1016/0014-5793(90)80175-i. [DOI] [PubMed] [Google Scholar]
- 40.Yang Q, Kadam A, Trempe JP. Mutational analysis of the adeno-associated virus rep gene. J Virol. 1992;66:6058–6069. doi: 10.1128/jvi.66.10.6058-6069.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker SL, Wonderling RS, Owens RA. Mutational analysis of the adeno-associated virus type 2 Rep68 protein helicase motifs. J Virol. 1997;71:6996–7004. doi: 10.1128/jvi.71.9.6996-7004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wonderling RS, Kyostio SR, Owens RA. A maltose-binding protein/adeno-associated virus Rep68 fusion protein has DNA-RNA helicase and ATPase activities. J Virol. 1995;69:3542–3548. doi: 10.1128/jvi.69.6.3542-3548.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith RH, Kotin RM. The Rep52 gene product of adeno-associated virus is a DNA helicase with 3′-to-5′ polarity. J Virol. 1998;72:4874–4881. doi: 10.1128/jvi.72.6.4874-4881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansilla-Soto J, Yoon-Robarts M, Rice WJ, Arya S, Escalante CR, Linden RM. DNA structure modulates the oligomerization properties of the AAV initiator protein Rep68. PLoS Pathog. 2009;5:e1000513. doi: 10.1371/journal.ppat.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarate-Perez F, Mansilla-Soto J, Bardelli M, Burgner JW, 2nd, Villamil-Jarauta M, Kekilli D, Samso M, Linden RM, Escalante CR. Oligomeric properties of adeno-associated virus Rep68 reflect its multifunctionality. Journal of virology. 2013;87:1232–1241. doi: 10.1128/JVI.02441-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zarate-Perez F, Bardelli M, Burgner JW, 2nd, Villamil-Jarauta M, Das K, Kekilli D, Mansilla-Soto J, Linden RM, Escalante CR. The interdomain linker of AAV-2 Rep68 is an integral part of its oligomerization domain: role of a conserved SF3 helicase residue in oligomerization. PLoS Pathog. 2012;8:e1002764. doi: 10.1371/journal.ppat.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maggin JE, James JA, Chappie JS, Dyda F, Hickman AB. The amino acid linker between the endonuclease and helicase domains of adeno-associated virus type 5 Rep plays a critical role in DNA-dependent oligomerization. Journal of virology. 2012;86:3337–3346. doi: 10.1128/JVI.06775-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 54.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 57.Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amaratunga M, Lohman TM. Escherichia coli rep helicase unwinds DNA by an active mechanism. Biochemistry. 1993;32:6815–6820. doi: 10.1021/bi00078a003. [DOI] [PubMed] [Google Scholar]
- 59.Zolotukhin S, Potter M, Hauswirth WW, Guy J, Muzyczka N. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J Virol. 1996;70:4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 61.Yoon M, Smith DH, Ward P, Medrano FJ, Aggarwal AK, Linden RM. Amino-terminal domain exchange redirects origin-specific interactions of adeno-associated virus rep78 in vitro. Journal of virology. 2001;75:3230–3239. doi: 10.1128/JVI.75.7.3230-3239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiorini JA, Afione S, Kotin RM. Adeno-associated virus (AAV) type 5 Rep protein cleaves a unique terminal resolution site compared with other AAV serotypes. J Virol. 1999;73:4293–4298. doi: 10.1128/jvi.73.5.4293-4298.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fogolari F, Corazza A, Yarra V, Jalaru A, Viglino P, Esposito G. Bluues: a program for the analysis of the electrostatic properties of proteins based on generalized Born radii. BMC Bioinf. 2012;13:S18. doi: 10.1186/1471-2105-13-S4-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hewitt FC, Samulski RJ. Creating a novel origin of replication through modulating DNA-protein interfaces. PLoS One. 2010;5:e8850. doi: 10.1371/journal.pone.0008850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hewitt FC, Li C, Gray SJ, Cockrell S, Washburn M, Samulski RJ. Reducing the risk of adeno-associated virus (AAV) vector mobilization with AAV type 5 vectors. Journal of virology. 2009;83:3919–3929. doi: 10.1128/JVI.02466-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franke D, Kikhney AG, Svergun DI. Automated acquisition and analysis of small angle X-ray scattering data. Nucl Instrum Methods Phys Res, Sect A. 2012;689:52–59. [Google Scholar]
- 67.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- 68.Tria G, Mertens HDT, Kachala M, Svergun DI. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015;2:207–217. doi: 10.1107/S205225251500202X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hickman AB, Dyda F. Binding and unwinding: SF3 viral helicases. Curr Opin Struct Biol. 2005;15:77–85. doi: 10.1016/j.sbi.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 70.Bardelli, M. e. a. Manuscript in preparation.
- 71.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang YP, Xu M, Machado AC, Yu XJ, Rohs R, Chen XS. Mechanism of origin DNA recognition and assembly of an initiator-helicase complex by SV40 large tumor antigen. Cell Rep. 2013;3:1117–1127. doi: 10.1016/j.celrep.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.