

Abstract
Although at the genetic level cancer is caused by diverse mutations, epigenetic modifications are characteristic of all cancers, from apparently normal precursor tissue to advanced metastatic disease, and these epigenetic modifications drive tumour cell heterogeneity. We propose a unifying model of cancer in which epigenetic dysregulation allows rapid selection for tumour cell survival at the expense of the host. Mechanisms involve both genetic mutations and epigenetic modifications that disrupt the function of genes that regulate the epigenome itself. Several exciting recent discoveries also point to a genome-scale disruption of the epigenome that involves large blocks of DNA hypomethylation, mutations of epigenetic modifier genes and alterations of heterochromatin in cancer (including large organized chromatin lysine modifications (LOCKs) and lamin-associated domains (LADs)), all of which increase epigenetic and gene expression plasticity. Our model suggests a new approach to cancer diagnosis and therapy that focuses on epigenetic dysregulation and has great potential for risk detection and chemoprevention.
Even before the discovery of epigenetic modifications in cancer, classical tumour biology suggested that generalized disruption of gene expression might underlie the key properties of unregulated tumour growth, invasion and metastasis. Perhaps the earliest person to recognize the importance of gene expression in cancer was Sidney Weinhouse, who described a generalized disruption of the biochemistry of cancer cells that was focused on isozymes that were primarily related to metabolism1. However, since the discovery of oncogene mutation in human tumours2, the principal focus of cancer genetics has been on mutations. We argue in this Opinion article that, although key mutational changes are necessary for the initiation of what we currently recognize as neoplastic growth and are likely to be required for escape from a cellular niche, epigenetic modifications also have a crucial role: these modifications allow rapid cellular selection in a changing environment, thus leading to a growth advantage for the tumour cells at the expense of the host. This view does not contradict and indeed collaborates with the genetic model, but it puts epigenetics at the very heart of cancer biology, from normal precursor cells at the sites where cancer arises, and through all stages of tumour progression, to advanced metastatic disease.
The first experiments on DNA methylation in human cancer, which compared samples of human colorectal cancer with matched normal mucosa isolated from the same patients, showed widespread hypomethylation involving approximately one-third of single-copy genes3. In response to the discovery of tumour suppressor genes4, later studies focused on identifying silenced genes as surrogates for mutation, beginning with the observation of promoter hypermethylation of RB1 by Horsthemke and colleagues5,6. During the 2000s, the maturation of microarrays and the advent of next-generation sequencing technologies in combination with the rise of data-driven discovery in biology have led to important new insights. These include the discovery of genome-wide loss of epigenetic stability, which is common across disparate tumour types. This seems to be the underlying mechanism for both the hypomethylation and the hypermethylation of individual genes, which was the historical focus of this field7. In addition, recently discovered mutations in the epigenetic apparatus probably contribute to epigenetic disruption in cancer. We review these recent discoveries and point to the possibility that cancer is a state in which the epigenome is allowed to have greater plasticity than it is supposed to have in normal somatic tissues. This increased epigenetic plasticity is a normal component of development or postnatal responses to injury, but its constitutive activation in cancer causes epigenetic heterogeneity that leads to most of the classical cancer hallmarks. We discuss below how this perspective provides new research avenues for diagnostics and treatment.
Large epigenetic structures
Just as the field of cancer epigenetics was presaged by early studies of abnormal gene expression, the role of large epigenetic structures in cancer was indicated by the earliest studies of cancer epigenetics by Theodor Boveri, who described abnormal chromatin in cancer cells in photomicrographs in 1929 (REF. 8). Alterations in nuclear shape are often used for diagnosis and are potentially symptomatic of the disorganization of this carefully regulated state9. In addition, nuclear lamina proteins (which serve to retain nuclear organization) show altered gene expression in cancer10. We describe below advances from whole-genome analyses that begin to provide molecular detail to these altered structures in cancer.
Chromatin LOCKs and LADs
Euchromatin refers to genes that are more open to transcription owing to post-translational modifications of histones and lower nucleosome density, whereas heterochromatin is the opposite: genes that are less open to transcription owing to greater nucleosome density and certain post-translational histone modifications. Typically, facultative heterochromatin — that is, a region that can switch between transcriptionally repressive states and activated states — is examined at a local gene level. However, in addition to small-scale changes, the genome is partitioned into large euchromatic and heterochromatic domains, which have been given different names for mostly overlapping structures by laboratories that have approached this organization using varying methods. We recently reported large organized chromatin lysine modifications (LOCKs), which are defined by genomic domains enriched for heterochromatin post-translational modifications, such as histone H3 lysine 9 dimethylation (H3K9me2)11. LOCKs expand during differentiation and are lost in cancer11 (FIG. 1a,b). Heterochromatic regions can also be defined by their organization and position within the nucleus: DNA sequences associated with proteins in the nuclear lamina are known as lamina-associated domains (LADs)12. Heterochromatic regions defined by histone modifications (LOCKs) and those defined by nuclear location (LADs) have been shown to have 80% overlap in different samples11,13,14, but a causal relationship, as in LADs controlling chromatin or chromatin informing nuclear location, has not yet been proved.
Figure 1. Alterations in the cancer epigenome that can cause epigenetic dysregulation.
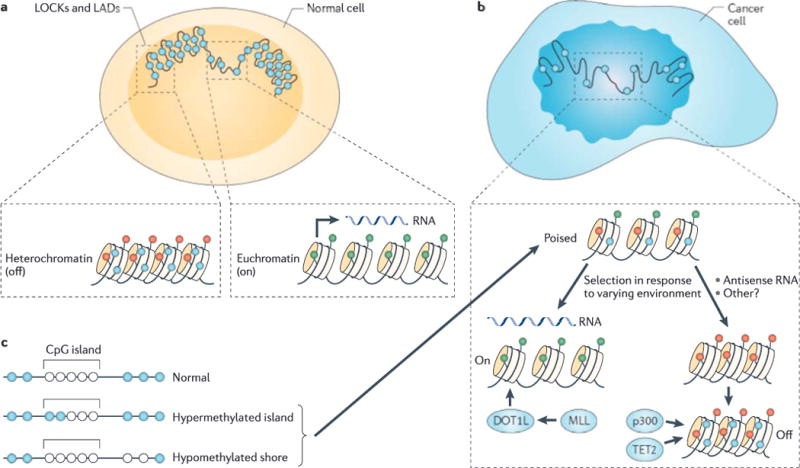
a | Large organized chromatin lysine modifications (LOCKs) and lamin-associated domains (LADs) (shown here in large scale) are associated with the nuclear membrane and are generally heterochromatic, with a high level of DNA methylation. Transcriptionally active genes have less compact nucleosomes than silent genes, and active and silent genes are distinguished by differing post-translational modifications of histones (green represents on and red represents off), as well as increased DNA methylation (shown in blue) of silent genes, b | In cancer there is a reduction of LOCKs, as well as general disorganization of the nuclear membrane and hypomethylation of large blocks of DNA corresponding approximately to the LOCKs and LADs. Chromatin is in a more stem cell-like state with the ability to differentiate into euchromatin and hypomethylated genes, or into heterochromatin and hypermethylated genes. Our argument is that epigenetic dysregulation allows for selection in response to the cellular environment for cellular growth advantage at the expense of the host. Mechanisms include mutations in epigenetic regulatory genes (for example, DOT1-like (DOT1L), mixed lineage leukaemia (MLL), EP300 (which encodes p300) and tet methylcytosine dioxygenase 2 (TET2)) and primary epigenetic modifications with positive feedback. c | Loss of boundary stability of methylation at CpG islands includes the encroaching of boundaries, leading to CpG island hypermethylation, and the shifting out of boundaries, leading to hypomethylated CpG shores. Both mean shifts in methylation and hypervariability allow for selection.
LOCKs and LADs change during development, generally increasing in size. Genes in LADs are typically transcriptionally repressed15, but by artificially reorganizing the nucleus to move genes to the nuclear periphery, transcription profiles and histone modifications of chromatin containing these genes are drastically altered15. Genes encoding proteins that are involved in organizing the nuclear membrane also have altered expression in many different cancer types16. Different laboratories have observed dynamic changes in chromatin state by examining different histone sites — for example, H3K9me2, H3K9 trimethylation (H3K9me3) or H3K27me3 — but still note that the prevalence of heterochromatic regions is associated with the differentiation state of the cell17,18. LOCKs are also altered in cells undergoing epithelial-mesenchymal transition (EMT), an important behaviour in cancer progression: during EMT, chromatin is reprogrammed in bulk, which results in a dramatic loss of H3K9me2 and an increase of H3K4me3 and H3K36me3 (REF. 19). Chromatin immunoprecipitation followed by microarray (ChIP-chip) experiments carried out on mouse chromosomes 4–14 showed loss of H3K9me2 in 96% of LOCKs but not in non-LOCK regions19.
The study of LOCKs and LADs in cancer is very new, and even chromatin modifications that are known targets of mutations, such as H3K27me3, have not yet been analysed systematically at a genome-scale sequencing level in cancer. A great deal of detail and mechanism needs to be fleshed out. For example, LOCKs and LADs may themselves be nuanced with regard to combinations of chromatin marks that define physiologically distinct domains20. To date, other than pilot studies, there has been no systematic analysis of primary human cancers and matched normal tissues with respect to LOCKs and LADs. More detailed study has been carried out on blocks of DNA methylation (discussed below), but these need to be related to LOCKs and LADs to form a complete picture of large-scale epigenetic alterations in cancer. Euchromatin islands provide a clue to a possible connection between LOCKs and LADs; these islands are small regions within the larger LOCKs and LADs that have reduced amounts of heterochromatin and are enriched for DNase hypersensitive sites and differentially methylated regions in cancer21.
Hypomethylated blocks
We recently made a surprising discovery by using whole-genome bisulphite sequencing of human colorectal cancer samples, and this finding helps to explain the earliest observation in cancer epigenetics: the widespread hypomethylation of genes in cancer3. By comparing three samples of colorectal cancer to matched normal mucosa from the same patients, we identified long blocks of hypomethylated DNA in cancer with a median size of 28 kb and a maximum size of 10Mb (a range of 5kb–10Mb)7 (FIG. 1a,b). In blocks, normal samples exhibited methylation levels of ~80%, and the cancer samples ranged from 40% to 60%. One-third of transcriptional start sites are contained within the large hypomethylated blocks. Furthermore, these hypomethylated blocks mostly corresponded to LOCKs and LADs, uncovering a surprising relationship between large nuclear domains of both DNA and chromatin that are disrupted in cancer7. These findings were subsequently confirmed by others22.
There is a point of confusion in the literature that we wish to clarify: in older literature based on Southern hybridization of long interspersed nuclear element (LINE) or Alu sequences, it seems that the DNA hypomethylation in cancer is due to repetitive sequences and not to single-copy genes23. However, modern whole-genome bisulphite sequencing methods have demonstrated that repetitive sequences, although somewhat enriched in hypomethylated blocks, are in fact no more hypomethylated than non-repetitive sequences in the blocks7.
What is the potential role of hypomethylated blocks in cancer? An intriguing suggestion comes from an analysis of gene expression. Although the overall level of gene expression in hypomethylated blocks remains low in cancer, the hypomethylated blocks contain the most variably expressed genes in tumours compared with normal controls7. Furthermore, the DNA methylation levels in these regions were not only reduced, they were also extremely variable in the quantitative levels of DNA methylation7. Thus, although mean changes in gene expression and DNA methylation in cancer are important, their heterogeneity may be equally or even more important in tumour heterogeneity and cancer progression and may underlie tumour cell heterogeneity.
Furthermore, similar structures — known as partially methylated domains (PMDs) — have been found to be relevant in differentiation and reprogramming. PMDs are large regions that are differentially methylated between embryonic stem cells and fibroblasts24, as well as between induced pluripotent stem (iPS) cells and fibroblasts25. These areas generally overlap with the blocks found in cancer7 and are hypomethylated in more differentiated cells. This reinforces the idea that there is a strong link between the epigenetic loci dysregulated in cancer and the loci that show controlled alteration in differentiation.
In addition, the hypomethylated blocks may contribute to mutation. Hypomethylated loci in cancer often coordinate with DNA-break hotspots, and may therefore contribute to copy number changes26. As these primary observations are so new, it is likely that additional mechanisms linking these considerable regional DNA methylation changes to cancer will be uncovered over time.
5-hydroxymethylcytosine is a proposed intermediate in the demethylation of cytosine, but it is indistinguishable from 5-methylcytosine by bisulphite- or restriction enzyme-based techniques27. Affinity-based methods (for example, 5-hydroxymethylcytosine DNA immunoprecipitation (hMeDIP) and methyl-CpG binding domain (MBD)-binding assays28) and chemical methods (for example, liquid chromatography-mass spectrometry (LC-MS)29) can distinguish hydroxymethyl from methyl modifications. Although the absolute level of hydroxymethylation in most normal tissue is low, recent work has shown a relative reduction in the levels of hydroxymethylation in melanoma30, liver cancer29 and colorectal cancer31. New single-base-resolution methods that are sensitive to hydroxymethylation are emerging to aid in distinguishing differences in hydroxymethylation from total methylation in cancer32,33.
Small epigenetic structures
The role of DNA methylation in smaller regions of DNA, such as CpG islands (CGIs), is part of the classical cancer epigenetics literature, but here too our perspective has been greatly changed by the advent of newer genomic technologies. For example, the existence of CpG island shores (CGI shores) and of asymmetric division of nucleosomes during DNA replication were unknown until recently.
CGIs and shores
In 1982, Wolf and Migeon34 discovered highly CpG-enriched sequences that, when methylated on the inactive X chromosome, are associated with silencing of housekeeping genes. Bird and colleagues35 later identified what they termed islands of CpG-rich sequences enriched at genes throughout the genome.
The observation of the hypomethylation of genes in human colorectal cancer3 was extended shortly thereafter to a larger series of tumours, including pre-malignant adenomas, with hypomethylation as an apparently ubiquitous feature of cancer36 (FIG. 1a,b). The overall global reduction of 5-methylcytosine in tumours was confirmed by quantitative high-performance liquid chromatography (HPLC)37. Many laboratories identified genes activated by hypomethylation, including oncogenes, such as HRAS38, and the families of genes expressed normally in testis and aberrantly activated in tumours, such as the melanoma-associated antigen (MAGE) family in melanoma39. Additional high-throughput array-based methods have identified hundreds of genes that are epigenetically activated in various cancers, including lung, gastric, colon, pancreatic, liver and cervical cancers40–48.
Arguing that epigenetic gene silencing might involve tumour suppressor genes, in 1991 Horsthemke5 and Dryja6 independently identified hypermethylation of a CGI upstream of the RB1 tumour suppressor gene5,6. Many tumour suppressor genes have since been associated with hypermethylated CGIs49 (FIG. 1a,b). However, there are several conundrums in this work. One issue is that much of this research was dedicated to the analysis of stable tumour cell lines and immortalized cell lines, which show marked hypermethylation of CGIs in general50. Furthermore, as Bestor and others51–53 have repeatedly pointed out, most hypermethylated tumour suppressor gene-associated CGIs are not in the promoters of these genes, and thus the hypermethylation of these sequences is likely to be consequential rather than causal51–53. It has been proposed that a priori methylation is a mechanism of tumour suppressor gene silencing that can cause cancer predisposition (rather than being a late event in tumorigenesis) in a similar manner to the cancer predisposition that is caused by germline mutations; however, the data supporting this have been relatively sparse51. The most exciting example is MLH1 methylation transmitted as a germline trait, but this report was repudiated by most of its authors owing to contamination of the germ cells with stroma54,55.
Indeed, we believe the mechanism of tumour suppressor gene silencing to be primarily driven by chromatin modification and not by DNA methylation. Vogelstein and colleagues56 showed in 2003 that tumour suppressor gene silencing seems to be driven by histone modifications before DNA methylation changes56. Recently, Sproul and colleagues57 directly showed that DNA hypermethylation of tumour suppressor genes in breast cancer occurs at sites that are already repressed in normal cells of the same lineage57. The same group extended these convincing results regarding the lack of a role for DNA hypermethylation in cancer development to 1,154 human cancer samples from seven different tissue types58. Similar findings at individual loci have also been shown by others52,53.
Loss of CGI boundary stability in cancer
With the advent of whole-genome epigenetic analysis, it was possible to broaden the focus of cancer epigenetics to consider regions outside the relatively limited CGIs. We designed a microarray using an algorithm that is agnostic to genes and CGIs59, and found that most methylation differences between tissues (tissue-specific differentially methylated regions (tDMRs)) occurred outside CGIs, often within 2kb of CGI boundaries in regions that are now commonly referred to as CGI shores60. A retrospective analysis of previous work agrees with this result; tDMR locations are more common in low-CpG-density promoters, although the connection to CGI shores was not identified24.
Furthermore, compared with the fibroblasts of origin, 70% of altered methylation regions (reprogramming-specific differentially methylated regions (rDMRs)) in iPS cells are located in CGI shores. This suggests that shores are important for differentiation and reprogramming61. Indeed, colon cancer can in most cases be distinguished from normal colon tissue using these rDMRs, suggesting that carcinogenesis may involve a partial reprogramming of the epigenome towards a more stem cell-like state61.
What is the function of CGI shores? One possibility is that they are sites of alternative transcription and enhancer binding regions; this is in contrast to CGIs, which Wolf and Migeon34 and others demonstrated are strongly protected from DNA methylation to maintain housekeeping gene function34. Indeed, hypomethylated CGI shores were shown to activate alternative transcriptional start sites proximate to cancer-specific differentially methylated regions (cDMRs), as shown by 5′ rapid amplification of cDNA ends (RACE) experiments60.
Another clue to the function of CGI shores comes from whole-genome bisulphite sequencing. Cancers lose the sharply demarcated boundary between high and low methylation that is defined by CGIs; that is, at the CGI shores7. Thus, when the boundary between high and low methylation shifts inwards towards the CGI, the CGI shore becomes hypermethylated. When the boundary shifts outwards, the CGI shore becomes hypomethylated (FIG. 1c). The erosion of these sharply defined boundaries results in altered gene expression7.
Furthermore, a striking hypervariability in DNA methylation is found at these CGI shores or boundaries in cancer samples, similar to the hypervariability of DNA methylation described above for the large blocks7 (FIG. 1c). This same property of hypervariable DNA methylation at the CGI shores or boundaries and blocks is a general property of cancer, affecting at least breast, colorectal, kidney, lung and thyroid tumours7. Tissue heterogeneity does not explain this hypervariability because normal tissue displays even greater heterogeneity than cancer samples7. In fact, an increase in methylation hypervariability in phenotypically normal tissue is predictive of future cancer development62. A similar hypervariability is found in gene expression in cancer63, and the most hypervariable of these genes are found within the large blocks7. We emphasize that mean changes in gene expression are important but that variance may be equally important in tumour progression.
Small chromatin domains
Individual mucleosomes, as opposed to the large domains described above, are also likely to be involved in affecting tumour progression. The organization of chromatin into euchromatic and heterochromatic structures is controlled by nucleosome positioning, which functions together with post-translational modifications of histone tails (for example, in enhancers)64,65. Physical access to DNA is restricted by nucleosome positioning and packing — chromatin remodelling complexes act to alter this in cancer66. For example, transcriptional activity is associated with nucleosome depletion67, with transcription-activating histone modifications such as acetylated H3K14 (REF. 68) and with the presence of specific histone variants such as H3.3 (REF. 69) and H2A.Z70. It is important to consider the complex combinatorial nature of the histone code; different histone modifications often act together, meaning that each modification must be considered in context with the other modifications that are present on the nucleosome71. Some regions are even bivalent, with nucleosomes having both H3K4me3 (a euchromatic, transcriptionally active modification) and H3K27me3 (a heterochromatic, repressive modification), implying a metastable pluripotent state72. Although previous work has demonstrated that bivalent modifications do not occur on the same histone tail73, more recent work has shown that these opposing modifications localize to a single nucleosome, with repressive and activating marks on different H3 proteins within the same nucleosome74. These same regions are associated with hypermethylated CGIs in cancer75,76 (FIG. 1a,b) and reprogramming61, and in fact a relationship has been shown between DNA methylation and nucleosome positioning77 and histone modification18.
We and others78,79 have identified one mechanism for heterochromatin-induced silencing of tumour suppressor genes. Antisense expression of cyclin-dependent kinase inhibitor 2B (Cdkn2b, which encodes p15) generates heterochromatin formation at the sense promoter, leading to gene silencing, and this mechanism seems to be important in leukaemia78,79. Hypermethylation ensues on cell differentiation, and the expression of antisense RNA has been shown to act as a mediator of chromatin remodelling and heterochromatin formation (FIG. 1a,b). Intriguingly, CGI hypermethylation does occur but only arises after heterochromatin formation79. Cis-acting non-coding RNAs at promoters and enhancers can regulate chromatin at gene promoters80. The observation of chromatin modifications preceding DNA methylation during differentiation resonates with the observations that CGI hypermethylation arises secondarily to chromatin modifications in tumour suppressor gene silencing56, and that the hypermethylated CGIs in cancer are located at genes that have already been silenced in normal tissue57,58, presumably through chromatin.
Mutations and the epigenome
The recent discovery of several mutated epigenetic modifiers in human cancer provides a potential mechanism by which DNA mutation might lead to epigenetic alterations. Given the apparently universal presence of DNA methylation and chromatin alterations in human cancer, we summarize below the frequency of mutations of epigenetic modifying genes, going gene by gene and tumour by tumour, beginning with the Catalogue of Somatic Mutations in Cancer (COSMIC) database81 and then reviewing the original citations. The classes of genes include histone variants (direct substitution of a mutant histone isoform); DNA methyltransferases; histone acetyltransferases; histone deacetylases; histone methyltransferases; histone demethylases; and chromatin remodelling factors, which can induce changes in euchromatin and heterochromatin (TABLE 1). Mutations in chromatin readers are also occasionally involved in cancer but apparently not as drivers of cancer progression82.
Table 1.
Epigenome-modifying gene mutations in human cancer
| Gene | Cancer | Frequency or stage of cancer | Frequency of mutation (N) | Effect | Refs |
|---|---|---|---|---|---|
| Histone variants | |||||
| HIST1H1B | Colorectal cancer | Common | 4% (24) | 149 | |
| HIST1H1C | Non-Hodgkin’s lymphoma | Common | 7% (127) | 150 | |
| H3F3A | Paediatric glioblastoma | Rare aggressive paediatric, high grade | 36% (90) | Prevents PTMs on H3K27 or H3K36 | 85 |
| Diffuse intrinsic pontine glioma | Rare aggressive paediatric | 60% (50) | Prevents PTMs on H3K27 | 95 | |
| HIST1H3B | Diffuse intrinsic pontine glioma | Rare aggressive paediatric | 18% (50) | Prevents PTMs on H3K27 | 95 |
| DNA methyltransferases | |||||
| DNMT1 | Colorectal cancer | 2% (29) | Mutation | 151 | |
| DNMT3A | AML | Stage M4 | 13.6% (66) | 87 | |
| Stage M5 | 20.5% (112) | 87 | |||
| AML | Common | 22.1% (281) | 88 | ||
| DNA demethylases | |||||
| TET2 | BCR-ABL-negative myeloproliferative neoplasms | Rare form | 13% (239) | 152 | |
| CMML | Common form | 50% (88) | 90 | ||
| MDS | Rare | 26% (102) | 153 | ||
| IDH1 | Anaplastic astrocytoma | Rare | 73% (52) | 154 | |
| Diffuse astrocytoma | Rare | 90% (30) | 154 | ||
| AML | Common | 6.2% (385) | 89 | ||
| IDH2 | AML | Common | 8.6% (385) | 89 | |
| Historie acetyltransferases | |||||
| EP300 (which encodes p300) | Pancreas adenocarcinoma | Common form | 8% (24) | Mutation | 83 |
| DLBCL | Common form | 10% (134) | Mutation | 91 | |
| Follicular lymphoma | Uncommon form | 8.7% (46) | Mutation | 91 | |
| Head and neck squamous cell cancer | Common | 11% (74) | Mutation | 155 | |
| Transitional cell carcinoma (bladder) | Common | 13% (97) | Mutation | 156 | |
| CREBBP (which encodes CBP) | Ovary | Common | 3% (75) | Inactivated | 157 |
| Breast adenocarcinoma | Common | 8% (183) | Gain of copy | 158 | |
| Lung cancer | Common | 5.3% (95) | Mutation | 159 | |
| DLBCL | Common form | 22.4% (134) | Mutation | 91 | |
| DLBCL | Common form | 18% (111) | Mutation | 92 | |
| Follicular lymphoma | Uncommon form | 32.6% (46) | Mutation | 91 | |
| Relapsed ALL | 18.3% (71) | Mutation | 94 | ||
| Transitional cell carcinoma (bladder) | Common | 13% (97) | Mutation | 156 | |
| ELP4 | Breast adenocarcinoma | Common | 4% (183) | Gain of copy | 158 |
| Historie deacetylases | |||||
| HDAC4 | Breast adenocarcinoma | Common | 4% (24) | Mutation | 149 |
| HDAC9 | Prostate adenocarcinoma | Common | 42.9% (7) | Mutation | 160 |
| Historie methyltransferases | |||||
| SETD2 | Renal clear cell carcinoma | Common | 3% (407) | Mutation | 161 |
| MEN1 | Pancreas neuroendocrine cancer | Rare | 44% (68) | Mutation | 84 |
| Parathyroid cancer | Rare | 35% (185) | Mutation | 162 | |
| MIL | Squamous cell lung cancer | Rare | 3% (63) | Mutation | 158 |
| Transitional cell carcinoma (bladder) | Common | 7% (97) | Mutation | 156 | |
| Mixed lineage leukaemia | Common | 100% (definition) | Fusion | 86 | |
| MLL2 | Renal clear cell carcinoma | Common | 4% (407) | Mutation | 161 |
| Childhood medulloblastoma | Rare | 8.7% (92) | Mutation | 163 | |
| Childhood medulloblastoma | Rare | 13.6% (88) | Mutation | 164 | |
| DLBCL | Common form | 32% (37) | Mutation | 150 | |
| DLBCL | Common form | 22.8% (92) | Mutation | 92 | |
| Follicular lymphoma | Uncommon form | 89% (35) | Mutation | 150 | |
| Head and neck squamous cell cancer | Common | 11% (74) | Mutation | 155 | |
| MLL3 | Childhood medulloblastoma | Rare | 3.4% (88) | Mutation | 164 |
| Transitional cell carcinoma (bladder) | Common | 5% (97) | Mutation | 156 | |
| Colorectal cancer | Common | 20.8% (24) | 149 | ||
| EZH2 | Non-Hodgkin’s lymphoma | Common | 7.8% (681) | Mutations | 165 |
| DLBCL | Common form | 5.6% (107) | Mutation | 92 | |
| MDS and MPNs | Rare | 12% (219) | Mutations | 166 | |
| Myelofibrosis | Rare | 13% (30) | Mutations | 166 | |
| Follicular lymphoma | Uncommon form | 12% (221) | Mutations | 167 | |
| Histone demethylases | |||||
| KDM5C (also known as JARID1C) | Renal clear cell carcinoma | Common | 3% (407) | Mutation | 161 |
| KDM6A (also known as UTX) | Transitional cell carcinoma (bladder) | Common | 20% (97) | Mutation | 156 |
| Childhood medulloblastoma | Rare | 3.2% (92) | Mutation | 163 | |
| KDM2B | DLBCL | Uncommon form | 7.4% (54) | Mutation | 92 |
| Chromatin remodelling factors | |||||
| ARID1A | Pancreas adenocarcinoma | Common | 8% (24) | Mutation | 83 |
| Ovarian clear cell carcinoma | Rare | 57% (42) | Mutation | 96 | |
| Ovarian clear cell carcinoma | Rare | 46% (119) | Mutation | 97 | |
| Endometrial cancer | Common | 30% (33) | Mutation | 97 | |
| Transitional cell carcinoma (bladder) | Common | 13% (97) | Mutation | 156 | |
| Hepatocellular carcinoma | Common | 16.8% (125) | Mutation | 168 | |
| Colorectal adenocarcinoma | Hypermutated | 37% (30) | Mutation | 169 | |
| Non-hypermutated | 5% (165) | ||||
| ARID1B | Breast adenocarcinoma | Common | 5% (100) | Mutation | 170 |
| ARID2 | Hepatocellular carcinoma | Common | 5.6% (125) | Mutation | 168 |
| Melanoma | Common | 9% (121) | Nonsense mutation | 171 | |
| CHD1 | Prostate adenocarcinoma | Common | 42.9% (7) | Mutation | 160 |
| CHD5 | Prostate adenocarcinoma | Common | 42.9% (7) | Mutation | 160 |
| PBRM1 | Clear cell renal carcinoma | Common | 41% (227) | Mutation | 172 |
| ATRX | Pancreas neuroendocrine cancer | Rare | 25% (68) | Mutation | 84 |
| DAXX | Pancreas neuroendocrine cancer | Rare | 17.6% (68) | Mutation | 84 |
| SMARCD1 | Breast adenocarcinoma | Common | 4% (100) | Mutation | 170 |
| SMARCB1 (also known as SNF5 and INI1) | Malignant rhabdoid cancer | Rare | 100% (29) | Loss of copy or mutation | 173 |
| SMARCA4 | Childhood medulloblastoma | Rare | 4.3% (92) | Mutation | 163 |
ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; ARID, AT-rich interactive domain; ATRX, α-thalassaemia/mental retardation syndrome X-linked; CHD, chromodomain helicase DNA binding protein; CMML, chronic myelomonocytic leukaemia; CREBBP, CREB binding protein; DAXX, death-domain associated protein; DLBCL diffuse large B cell lymphoma; DNMT, DNA methyltransferase; ELP4, elongator acetyltransferase complex subunit 4; EZH2, enhancer of zest homologue 2; H3F3A, histone H3 family 3A; HDAC, histone deacetylase; HIST1H1B, histone cluster 1, H1b; HIST1H1C, histone cluster 1, H1c; HIST1H3B, histone cluster 1, H3b; IDH, isocitrate dehydrogenase; KDM, lysine-specific demethylase; MDS, myelodysplastic syndrome; MEN1, multiple endocrine neoplasia I; MLL, mixed lineage leukaemia; MPNs, myeloproliferative neoplasms; PBRM1, polybromo 1; PTMs, post-translational modifications; SETD2, SET domain containing 2; TET2, tet methylcytosine dioxygenase 2.
Our analysis of the mutation frequency of epigenome modifiers in cancer reveals a surprising pattern. Although there is clearly a relationship between mutations and epigenetic modification in cancer, most of the epigenetic-associated mutations in solid tumours identified to date involve either rare aggressive variants of adult tumours or paediatric cancers. For example, the common form of pancreatic adenocarcinoma shows an 8% mutation frequency in the histone acetyltransferase p300 (EP300)83, but the rarer pancreas neuroendocrine cancer has a 44% frequency of the histone methyltransferase multiple endocrine neoplasia I (MEN1)84 (TABLE 1). Similarly, childhood glioblastoma, an extremely rare brain cancer, shows frequent (35.6%) mutations in histone H3 family 3A (H3F3A), but adult glioblastomas show a drastically lower frequency of H3F3A mutations (3.4%)85 (TABLE 1).
By contrast, haematological cancers frequently involve chromosomal rearrangements of epigenetic modifiers, and this has been known for many years; for example, in mixed-lineage leukaemias86. Acute myeloid leukaemia involves mutations in several genes encoding proteins that modify DNA methylation, including DNA methyltransferase 3A (DNMT3A), isocitrate dehydrogenase 1 (IDH1) and IDH2. These mutations lead to either decreases in DNA methylation (IDH1 and IDH2 mutations) or increases in DNA methylation (DNMT3 A mutations)87–89. Chronic myelomonocytic leukaemia involves mutations in tet methylcytosine dioxygenase 2 (TET2), which is involved in DNA demethylation90. Lymphomas involve frequent inactivating mutations in the histone acetyltransferases EP300 and CREB binding protein (CBP; also known as CREBBP), leading to increased heterochromatin and resulting in gene silencing91,92. The histone methyltransferase mixed-lineage leukaemia (MLL), which undergoes translocations as a defining characteristic of mixed-lineage leukaemia, functions through DOTl-like (DOT1L), an H3K79me2 methyltransferase, which leads to specific gene activation93.
Epigenetic mutations also frequently occur in cancers that relapse or that are otherwise resistant to therapy, such as mutations of CBP in relapsed ALL94. A detailed analysis of these mutations can be found in TABLE 1. Chromatin remodelling proteins — for example, AT-rich interactive domain-containing protein 1A (ARID 1A) — are perhaps the most frequently mutated class of epigenetic modifying proteins in common solid tumours, and their consequent inactivation leads to increased levels of euchromatin and gene activation (TABLE 1). A dramatic example of epigenetic gene mutation coupled with aggressiveness in cancer is the recent finding that the histone H3 variant H3.3 is itself mutated in paediatric glioblastoma, thus preventing H3.3K27 modifications85,95. Such a mutation would be expected to substantially affect chromatin structure, causing aberrant gene expression and potentially allowing for the acquisition of aggressive properties; it should be noted that adult glioblastomas do not frequently contain this mutation. A similar argument can be made for ovarian clear cell carcinoma — mutations in the chromatin remodeller ARID1A were found to be common in this aggressive subtype in two different studies (57% and 46% frequency)96,97 but were not present in high-grade serous ovarian carcinoma97.
A very important possibility in considering the relationship between DNA mutations and epigenetic modification in cancer is that altered DNA methylation or chromatin modifications may change the mutation rate itself. For example, guanine quadruplexes (G4s) increase the risk of DNA breakage and activation of the homologous recombination DNA repair pathway; these breaks are inhibited by DNA methylation. Hypomethylated loci in cancer often coordinate with DNA break hotspots, and these are enriched in G4s26. Methylation-mediated mutation through spontaneous deamination may also give rise to mutation; 18.2% of inherited gene mutations occur as C–G>T–A mutations in CpG dinucleotides98; C–G>T–A mutations also make up the bulk of substitutions in many cancers99, even specifically at CpG dinucleotides in some cases83. More substantially, chromatin state has been shown to correlate extremely well with somatic mutation rate: H3K9me3 levels alone are predictive of >40% of somatic mutation loci in human cancer samples100. Common DNA fragile sites, which are implicated in copy number variation in cancer, also have decreased stability in regions of heterochromatin101. The organization of chromatin and genetic architecture of the nucleus have a direct effect on the rate and effectiveness of copy number alterations and rearrangements in cancer102. These data represent clear correlations between areas of epigenetic dysregulation and mutation, suggesting a collaborative effort between epimutation and genetic mutation in cancer development.
Abnormal expression of epigenetic modifier genes
It is also important to note that many alterations in the expression of epigenetic modifiers have been reported, and some of these seem to have an important role in tumour progression (and have been reviewed extensively elsewhere64,65,103). Perhaps the most notable example is overexpression of enhancer of zeste homologue 2 (EZH2), which results in increased H3K27me3 levels and thus silences tumour suppressor gene expression and promotes metastasis104. Therefore, these alterations in the epigenetic machinery are more complex to understand, and they should be viewed not as equivalent to the mutations summarized in TABLE 1 but rather as members of a positive feedback loop that leads to epigenetic dysregulation. An example of a positive epigenetic feedback loop in malignant transformation has recently been demonstrated directly in the nuclear factor-κB (NF-κB) pathway105. Another example is the class of reprogramming factors that lead to the generation of iPS cells106. We have also observed a significant overlap of rDMRs and cDMRs61. Furthermore, many of the reprogramming genes are overexpressed in cancer107. A non-exhaustive list of examples of alterations in gene expression of epigenetic modifiers in cancer is provided in TABLE 2.
Table 2.
Altered expression of some epigenetic modifying genes in cancer
| Gene | Change | Cancer | Refs |
|---|---|---|---|
| IGF2 | Increased | LOI in colorectal, gastric and breast cancers | 123,174,175 |
| Class 1 HDACs | Increased | Gastrointestinal, prostate, breast and cervical cancers | 176–180 |
| EZH2 | Increased | Prostate cancer | 104 |
| EZH2 | Increased | Breast cancer | 181 |
| HDACs | Increased | Several | 182 |
| HATs | Decreased | Several | 182 |
| HDACs | Increased | Colon cancer | 183,184 |
| HDAC6 | Increased | Breast cancer | 185 |
| SIRT1 | Increased | Prostate cancer | 186 |
| SIRT3 | Increased | Breast cancer | 187 |
| KDM5C | Increased | Breast cancer | 188 |
| SMYD3 | Increased | Liver, colon and breast cancers | 189 |
| EHMT1 | Decreased | Medulloblastoma | 190 |
| DNMT1 | Increased | Pancreas, liver, bladder and breast cancers | 191–194 |
| DNMT3B | Increased | Breast cancer | 195 |
| AID | Increased | Leukaemia | 196 |
AID, activation-induced cytidine deaminase; DNMT, DNA methyltransferase; EHMT1, euchromatic histone-lysine N-methyltransferase 1; EZH2, enhancer of zeste homologue 2; HAT, histone acetyltransferase; HDAC, histone deacetylase; IGF2, insulin-like growth factor 2; KDM5C, lysine-specific demethylase 5C (also known as JARID1B); LOI, loss of imprinting; SIRT, sirtuin; SMYD3, SET and MYND domain-containing 3.
One lesson from examining the expression of epigenetic modifiers in cancer is that the balance of euchromatic and heterochromatic histone modifications is crucial — a modification too far in either direction towards euchromatin or heterochromatin leads to dysregulation of gene expression and is advantageous for tumour growth (see REF. 108 for an example). This imbalance could be a target for therapy: for example, histone modifications could be brought back into balance through small-molecule inhibitors of histone deacetylases, histone acetyltransferases or histone methyltransferases108.
Cancer as epigenetic dysregulation
In summary, most cancers may share several common epigenetic modifications: large-scale alterations in chromatin involving LOCKs or LADs and hypomethylated blocks, and loss of methylated boundary stability at CGIs leading to hypermethylated CGIs, hypomethylated CGI shores and aberrant gene expression. This would lead to a drift towards a hybrid stem-somatic cell state, with increased methylation of Polycomb target regions and loss of methylation at pluripotency loci. A simple unifying explanation of these results is that cancer is caused by epigenetic dysregulation, which could account for the high degree of phenotypic variability that is observed among individual cancers and that leads to selection for cancer cell survival independent of the host.
To visualize this process, consider the classic ‘epigenetic landscape’ described by Waddington109, by which the normal epigenetic signature of the cell is represented by a ball trapped in a valley, the walls of which represent a restoring force constraining the ball in its normal state (FIG. 2a). Although this signature differs among tissues, it is highly regulated, invariant among individuals and ultimately defined genetically, according to the classic view109. In order to be dynamic and flexible, the epigenetic signature must allow for variation, provided in the form of intrinsic noise: that is, a biochemical characteristic of the system that leads to random departure from a set point110. For example, methylation inheritance shows an error rate that is estimated to be 4% for a given CpG motif per cell division in a cell population111. This is also consistent with the idea that epigenetic variability can lead to phenotypic selection on a much shorter timescale than can mutation112. Furthermore, variation in the epigenome may be controlled by factors in the genetic code, providing a potential mechanism for control of the level of this variation (which is represented by the slope of the valley walls)113.
Figure 2. Modelling epigenetic dysregulation using an Ornstein-Uhlenbeck process.
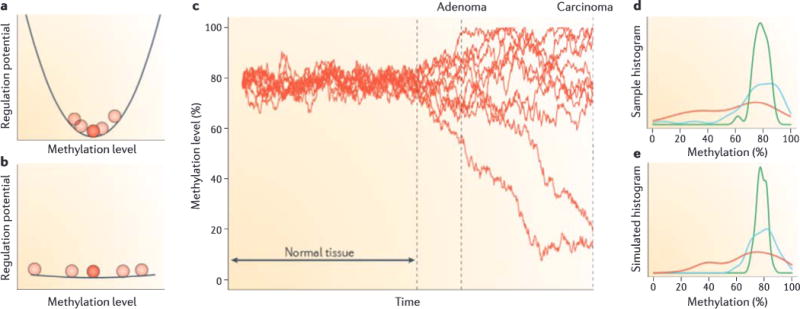
We used an Ornstein-Uhlenbeck process to model stochastic change in DNA methylation opposed by regulatory proteins. a | In normal tissue, the methylation level can be represented as a ball at the bottom of a valley—stochastic noise allows it to vary slightly, but regulatory forces represented by the walls of the valley keep the levels clustered around a single point, b | An early carcinogenic event flattens the landscape, leading to more variable methylation levels, c | Using the Euler-Murayama method, we can model this behaviour as = θ (μ—M)dt+σ dW, with M being the methylation value, μ the equilibrium point, θ the restoring force, σ the noise level (4%) and dW a Wiener process increment. Shown are ten example traces of simulated methylation levels. Regulatory forces (θ) are set high in the normal tissue and low after a carcinogenic event. As time progresses, samples of the simulation are taken, representing different stages of cancer progression: that is, normal, adenoma and carcinoma. d | Density plots of methylation data of a single CpG site from REF. 7 (Gene Expression Omnibus number: GSE29381) showing methylation histograms for normal (green; N = 29), adenoma (blue; N = 31) and carcinoma (red; N = 10) colon samples. The plots were generated by Gaussian kernel smoothed density function in R.e | Density plots of simulations for the same CpG showing the combined results from 100 simulations using the same sampling methods as in part d. The model provides an excellent fit to the data.
In cancer, an initial dysregulation of the epigenome would flatten this valley, breaking the delicate balance of regulation that maintained the stable epigenetic signature in the face of noise (FIG. 2b). This could be the result of repeated restructuring of the epigenetic landscape through inflammatory insult, as in the case of Barrett’s oesophagus114, or it could be through an initial mutation in one of the genes directing the fragile balance of the epigenome (almost any of the mutations presented in TABLE 1). A splicing variation in an epigenetic modifier gene could also cause dysregulation. For example, an isoform of DNMT3B commonly found in cancer, DNMT3B7, results in increased variation in the methylation signature115. Even the classical ‘gatekeeper’ mutations may have a role in unleashing epigenetic variation, given that cancer-associated mutants of adenomatous polyposis coli (APC), BRCA and p53 interact with epigenome-controlling proteins. We propose that after regulation of the epigenetic signature is relaxed, stochastic variation becomes the driving force in patterning the epigenome, allowing DNA methylation or chromatin structure to gradually diffuse away from its initial state. Natural selection within the host then allows each cancer to move differently, also leading to substantial epigenetic variation between a given cancer type across individuals or between metastases of a given cancer.
This epigenetic signature is constantly buffeted by stochastic variation, as if the ball is rocking randomly from side to side. The restoring force — that is, the network of genes that maintain epigenetic homeostasis — prevents the epigenetic signature from wandering too far from its equilibrium point in normal tissue. We can model this variation using an Ornstein-Uhlenbeck process (FIG. 2c); such processes are already used in biology for modelling selection pressure versus random genetic drift116. On the basis of this model, our argument that cancer involves a loss of regulation of the epigenome means that the restoring force is reduced or lost altogether (the positive feedback referred to above).
We generated simulated methylation levels using this process (FIG. 2c). Before carcinogenesis, methylation levels have relatively low variability, oscillating stochastically around their equilibrium point. When the simulation reaches the carcinogenesis point, we reduced the restoring force in the process, flattening the Waddington valley (FIG. 2b), and allowed the simulation to continue. The simulated methylation levels subsequently exhibited a random walk away from their previously well-ordered profile. It is important to note that the distance from the original equilibrium point increases over time, but not directionally: instead it increases by a diffusive spreading of the epigenetic signature (FIG. 2c).
We then applied our model to existing DNA methylation data from our previous work7, selecting a CpG from within a hypomethylated block as an example. The density plot of methylation values for normal, adenoma and cancer tissue at this CpG demonstrate the tight distribution in normal tissue, with a progressive relaxation from normal to adenoma to carcinoma (FIG. 2d). The simulated values match the actual data extremely well (FIG. 2d,e). This model is intended to suggest the underlying behaviour that may explain the increased distance from the normal profile we observe in tumour cells over time. We have not studied this exhaustively, and only one CpG is used as an example of the type of stochastic process that might apply. There are two interesting implications of this suggested model. First, the increased variation over time shown in data and matched by the model supports the idea that disruption of epigenetic regulation occurs at the earliest stages of cancer. Second, it shows that the idea of looking for a defined epigenetic signature for cancer is flawed. Rather, we should be looking for an anti-profile: that is, stochastic departure from a normal epigenetic signature.
Epigenetics and cancer hallmarks
Cancer is usually viewed as a complex group of multiple disorders that are mostly driven by somatic mutation and that involve the accretion of ten proposed properties: enhanced proliferation, growth suppressor evasion, anti-apoptosis, replicative immortality, angiogenesis, inflammation, altered metabolism, genomic instability and metastasis signalling117. We suggest that epigenetic disruption lies at the heart of all of these processes and that mutations enable and collaborate in these disruptions. The hallmark properties of cancer arise not only by mutation but also generally through stochastic epigenetic variation (as described above) and by natural selection of phenotypes that are advantageous to cancer cell survival and growth at the expense of the host. This is described graphically in FIG. 3, in which the epigenome sits at the intersection of the environment, genetic mutation and tumour cell growth.
Figure 3. Collaboration of epigenetic modification and mutation in the hallmarks of cancer.
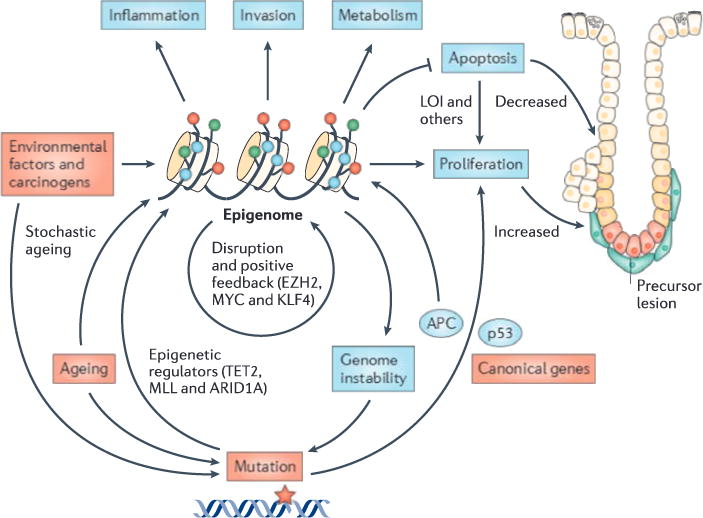
The epigenome sits at the intersection of the environment, genetic mutation and tumour cell growth. Environmental factors, such as carcinogens or diet, as well as injury and inflammation, cause epigenetic reprogramming. The epigenome also accumulates damage stochastically and through ageing. The machinery for maintaining epigenetic integrity can be stably disrupted in either of two ways: by mutation or by epigenetic change itself with positive feedback. Examples of mutation include epigenetic regulator mutations (TABLE 1), whereas examples of epigenetic change include loss of imprinting (LOI) of insulin-like growth factor 2 (IGF2) in colorectal carcinogenesis, enhancer of zeste homologue 2 (EZH2) silencing in prostate cancer (TABLE 2) and overexpression of reprogramming factors. The disruption of epigenetic integrity maintenance leads to the loss of epigenetic regulation and stochastic drift from a normal set point, followed by selection for cellular growth at the expense of other cells (FIGS 1,2). Some epigenetic modifications, such as shifting methylation boundaries at CpG islands and shores, lead to metabolic change and enhanced proliferation. Others, such as hypomethylated blocks, lead to increased invasion. Still others, such as LOI, directly change the balance between apoptosis and proliferation. Canonical mutations, such as in adenomatous polyposis coli (APC) and TP53 (which encodes p53), directly affect cancer hallmarks but can also cause epigenetic dysregulation. Similarly, epigenetic disruption, such as regional hypomethylation or CpG hypermethylation, can lead to increased chromosomal rearrangements and mutations, respectively. Instability of CpG island methylation boundaries also contributes to epigenetic dysregulation, allowing for selection in response to the cellular environment for cellular growth advantage at the expense of the host. ARID1A, AT-rich interactive domain-containing protein 1A; KLF4, Krüppel-like factor 4; MLL, mixed lineage leukaemia; TET2, tet methylcytosine dioxygenase 2.
Epigenetic damage arises from carcinogens or diet (for example, methionine), as well as injury and inflammation (for example, altered LOCKs and LADs in EMT, as described above). Errors in the maintenance of the epigenome over time — that is, simply ageing — may also have a role in accumulating stochastic epigenetic damage. The mechanism for maintaining epigenetic integrity is damaged in either of two ways: through mutation in the genes that encode these mechanisms (TABLE 1) or through epigenetic modifications themselves in such genes (TABLE 2) resulting in positive feedback. Examples of epigenetic modifications that lead to positive feedback include epigenetic silencing of EZH2 and loss of imprinting (LOI) in colorectal cancer. This damaged epigenetic maintenance machinery leads to stochastic drift from a normal epigenetic set point (FIG. 2) and selection for cancer hallmarks that give the cell a growth advantage at the expense of the host.
Some epigenetic modifications directly affect the hallmark properties of cancer; for example, shifts in the methylation boundaries at CGIs and CGI shores result in enhanced proliferation and metabolic change, and hypomethylated blocks lead to increased invasive potential61. In addition, shifting DNA methylation at CGI boundaries leads to hypomethylated shores and the activation of cell cycle genes that are overexpressed in cancer7, another cancer hallmark. In this case loss of DNA methylation, which seems to be a ground state for embryonic stem cells24, seems to promote gene expression, normally in development and abnormally in cancer. Similarly, LOI can directly change the balance between apoptosis and proliferation118,119 (FIG. 3).
Epigenetic dysregulation occurs very early in cancer. For example, DNA methylation is altered in the normal tissue of cancer patients, and these changes increase with age, suggesting a mechanism by which cancer frequency increases in an age-dependent manner120–122 (FIG. 3). A second example is LOI of insulin-like growth factor 2 (IGF2) in colorectal cancer, in which the normally silent allele becomes activated, leading to a double dose of this mitogen. In colorectal cancer, LOI of IGF2 is found in both the tumours and the normal tissue of affected patients123, and induced LOI of Igf2 in mice doubles the frequency of tumour development124. A third example is that Barrett’s oesophagus shows epigenetic modifications long before progression to overt malignancy114. Epigenetic modifications, particularly increased heterogeneous outlier variability in DNA methylation, are also predictive of malignant change in cervical cancer125.
Where do mutations fit into this process? We have previously suggested that evolution may favour mechanisms through genetic selection that allow for epigenetic heterogeneity by providing a selective advantage in a changing environment113. We suggest a similar mechanism might occur in cancer: that is, selection for epigenetic plasticity itself. More generally, we suggest that the initial dysregulation of the epigenome collaborates with crucial mutations to provide the phenotypic variation that allows the selection of the hallmark properties of cancer. There is interplay between epigenetic dysregulation, which provides the phenotypic heterogeneity that generates the hallmark properties of cancer and that potentially alters the mutation rate, and genetic mutations, which directly alter genes and pathways to confer hallmark properties of cancer, as well as enabling and assisting in epigenetic dysregulation (FIG. 3). Furthermore, the epigenetic changes themselves may contribute to increased mutation frequency.
We note that these mutations in epigenetic controlling genes generally occur in two types of tumours: haematopoietic malignancies and rare solid tumours (childhood solid tumours or aggressive subtypes of common adult solid tumours). The implication of this observation, which seems fairly well supported by a great deal of sequencing data, is that primary epigenetic modifications are a more prominent mechanism for cancer progression in common solid tumours than are mutations in epigenetic modifiers. The converse of this argument is that mutations in epigenetic modifiers have extremely strong effects on cellular behaviour, which is consistent with the profound aggressiveness of such tumours and their relative rarity. Haematopoietic malignancies are consistent with this view, as they commonly exhibit mutations in epigenetic modifier genes but arise almost completely progressed (that is, widely disseminated) compared with solid tumours. Even lymphomas, which can be relatively indolent for long periods, are more aggressive when associated with mutations in epigenetic modifiers.
Note that we do not exclude a primary role for mutations in conferring the hallmark properties of cancer, and in fact we believe that they do. We simply suggest that the epigenome is not relegated to merely a surrogate for mutation but rather that it has important non-local ramifications. Limiting our understanding of epigenetics in the context of cancer to the gene-centric view may neglect valuable insights into how cancer causes a general disruption of genetic regulation through the epigenome. With that in mind, even genes that are not normally thought to have a primary epigenetic role may have a strong interaction with the epigenome. For example, BRCA2, which at one time was thought to be a histone acetyltransferase126, is actually a binding partner of the histone acetyltransferase PCAF (also known as KAT2B)127. SMAD4 is mutated in pancreas adenocarcinoma and has been shown to be an important driver of this cancer type, but it also interacts with chromatin remodellers83. p53 directly interacts with DNMT1, which act together to control the expression of anti-apoptotic genes128. The epigenome can also be indirectly affected through pathways that are commonly mutated in cancer that can affect the expression level of epigenetic controlling proteins. Examples include the regulation of histone deacteylase 2 (HDAC2)129 and DNMT1 (REF. 130) by mutations of the WNT-β-catenin pathway, such as APC mutations in colorectal cancer. In fact, the protein stability of DNMT1, rather than its expression, may be altered by the PI3K-AKT pathway so that levels of the DNMT1 protein (and not mRNA) are changed without new expression131.
Implications for diagnosis and therapy
Viewing epigenetic dysregulation itself as a common driver of cancer progression has important implications for cancer diagnosis and therapy. For diagnosis it suggests a promising approach for identifying patients very early in the course of disease, and for therapy it suggests novel targets that could be the focus of therapeutic intervention early or even before the development of overt cancer.
In epigenetic detection, a great deal of effort has been invested in identifying CGI hypermethylation, with limited success49. We suggest that this is because an implication of the recent whole-genome epigenetic work shows that departure from a normal profile is more pathognomonic than a specific cancer epigenetic signature. For example, septin 9 (SEPT9) was reported to be a sensitive and specific serum-based marker for colorectal cancer132. However, a case-control study carried out independently of the commercial developer showed 90% sensitivity and 88% specificity for colorectal cancer but only 71% sensitivity for early cancers and 12% sensitivity for adenomas133, and therefore this marker is not useful for early screening. Similarly, methylation of bone morphogenetic protein 3 (BMP3), eyes absent homologue 2 (EYA2), ALX homeobox 4 (ALX4) or vimentin (VIM) was found in 66%, 66%, 68% and 72%, respectively, of primary colorectal cancers but in 7%, 5%, 11% and 11%, respectively, of normal mucosa134. One notable exception to the disappointing history of epigenetic detection of cancer is glutathione S-transferase pi 1 (GSTP1) in prostate cancer135, and this is probably because this particular gene has a crucial role in the early stage of the disease rather than being an indicator of epigenetic dysregulation per se.
If a detection scheme was developed on the basis of anti-profiling instead, a much higher sensitivity and predictive value might be achieved. Consistent with this idea, Teschendorff and colleagues125,136 recently showed that the DNA methylation variability was more predictive of cancer progression than mean changes in DNA methylation in both cervical125 and breast136 cancers. This test can differentiate between normal and neoplastic cervical tissue with 95% sensitivity and 78% specificity125. In fact, this test is predictive using tissue taken before the development of cervical neoplasia; neoplasia development within 3 years of sample collection is predictable with a 71% sensitivity and 50% specificity125. DNA methylation instability at specific loci is predictive of survival in endometrial, ovarian, cervical and breast cancers, emphasizing the usefulness of epigenetic instability in chemoresistance136. Such an approach could even be used in screening patients for early cancer: epigenetic anti-profiling of tumour cells using a sophisticated digital PCR-based approach has already shown considerable utility for the early diagnosis of colorectal cancer in stool137.
The methylation signature at specific CGIs can also suggest specific forms of tailored therapy, as in O6-methylguanine DNA alkyltransferase (MGMT) methylation, which can indicate the therapeutic choice for glioma138. However, other hypermethylated CGI markers have not shown a high degree of sensitivity or specificity for cancer or for key phenotypes, such as drug resistance, although such efforts are still ongoing139. In these cases DNA methylation is being used as a surrogate measure of gene expression and/or gene regulation.
In the case of haematological malignancies, in which mutations in epigenetic regulators are most frequent (TABLE 1), therapy targeted towards these pathways is already quite promising, including EZH2 inhibitors for lymphoma140,141, inhibitors of mutant IDH1 for acute myeloid leukaemia and myelodysplasia142, and HDAC inhibitors for cutaneous T cell lymphoma108. Additionally, a clever approach has been taken to inhibiting DOT1L, which is not mutated itself but which is a common target of MLL translocations143.
In our view, the most exciting potential application of this model of epigenetic dysregulation as a common driver throughout cancer progression is the potential for targeted chemoprevention. Currently, chemoprevention involves either nutritional recommendations or the use of non-prescription agents such as cyclooxygenase 2 (COX2; also known as PTGS2) inhibitors targeted towards individuals with a high risk of developing colorectal cancer, such as those with a strong family history of colorectal cancer144,145. The approach to chemoprevention is far more limited than in cardiology, in which chemoprevention with prescription medication (statins) is extremely effective at preventing heart disease146; this was a controversial approach when statins were first introduced for widespread use in the population. But what if we could identify the epigenetic disruption in patients before neoplastic growth even begins (as has been shown for cervical cancer125)? Then we might treat such patients with specific inhibitors even before they develop cancer. Similarly, LOI of IGF2 is associated with an increased frequency of colorectal cancer123 and may be associated with gastric cancer risk147. LOI of IGF2 also substantially increases the frequency of colon preneoplastic aberrant crypt foci in mice treated with the carcinogen azoxymethane, and inhibition of signalling at the IGF2 receptor reduces the incidence of neoplasms to a level even lower than that found in mice with normal Igf2 imprinting148. Thus, we might be able to identify disruption of the epigenome or even the risk of such disruption through epigenetic or genetic testing before cancer arises, and then treat patients preventively to reduce cancer incidence. This seems to us to be a potentially far more effective mechanism for reducing cancer mortality than the treatment of late-stage disease, and it would argue strongly for an epigenome-centred approach.
Acknowledgments
This work was supported by US National Institute of Health (NIH) grants CA05438 and HG03233 to A.P.F. The authors thank D. Singer, I. Ernberg and J. Bradner for helpful discussions.
Glossary
- Bivalent modifications
Nucleosomes containing both euchromatic histone H3 lysine 4 trimethylation (H3K4me3) and heterochromatic H3K27me3 post-translational modifications.
- Cancer hallmarks
Ten biological properties of cancer that are said to define the disease we argue that they arise by natural selection for cellular survival at the expense of the host in the setting of epigenetic dysregulation and random variation.
- Cancer-specific differentially methylated regions
(cDMRs). Differentially methylated regions that distinguish cancer cells from normal cells.
- Chemoprevention
Administration of pharmacological compounds to reduce cancer incidence without certain knowledge of its effect on a given patient.
- CpG islands
(CGIs). Areas of high CpG dinucleotide density in the genome, typically defined as a region at least 200 bp long with > 50% GC dinucleotides and an observed-to-expected CpG ratio of > 0.6.
- CpG island shores
(CGI shores). The region 2 kb on either side of a CpG island, and the location of most cancer-specific, tissue-specific and reprogramming-specific differentially methylated regions.
- Epigenetic dysregulation
The loss of normal control of DNA methylation or chromatin as a result of injury, epigenetic change or mutation, leading to phenotypic drift.
- Epigenetic variability
Increased inter-sample variation in the methylation or chromatin state. This was recently identified as a common property of cancer, allowing for more accurate detection between samples.
- Euchromatin
Areas of the genome that are more open to transcription owing to post-translational modifications of histones and with less nucleosome density.
- Heterochromatin
Areas of the genome that are less open to transcription owing to post-translational modifications of histones and with greater nucleosome density. Facultative heterochromatin can change between the two states. Large organized chromatin lysine modifications and lamina-associated domains describe heterochromatin over relatively large regions and are associated with the nuclear membrane.
- Hypomethylated blocks
Large (mean 144 kb) regions that are broadly hypomethylated in cancer and that mostly overlap with large organized chromatin lysine modifications and lamina-associated domains.
- Lamina-associated domains
(LADs). Genomic regions located in the nuclear periphery that are associated with lamina (an inner nuclear membrane-associated protein) and usually have low expression levels.
- Large organized chromatin lysine modifications
(LOCKs). Large heterochromatic regions characterized by low gene expression that are altered between somatic and stem cells; they are typically lost in cancer cells.
- Loss of imprinting
(LOI). Loss of parent of origin-specific expression in cancer of imprinted genes, first observed for insulin-like growth factor 2 (IGF2) in Wilms’ tumour and colorectal cancer.
- Ornstein–Uhlenbeck process
An overdamped Brownian harmonic oscillator — that is, stochastic variation from a normal state with no persistence of the rate of change — opposed by a stronger restoring force towards the equilibrium point. We are using this to model stochastic change in DNA methylation.
- Reprogramming-specific differentially methylated regions
(rDMRs). Differentially methylated regions that distinguish reprogrammed stem cells from somatic cells.
- Tissue-specific differentially methylated regions
(tDMRs). Differentially methylated regions that distinguish normal tissues from each other.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Winston Timp, Center for Epigenetics, Johns Hopkins University School of Medicine, 855N. Wolfe Street, Rangos 570, Baltimore, Maryland 21205, USA; Department of Biomedical Engineering, Johns Hopkins University School of Medicine, 720 Rutland Avenue, Baltimore, Maryland 21205, USA.
Andrew P. Feinberg, Center for Epigenetics, Johns Hopkins University School of Medicine, 855N. Wolfe Street, Rangos 570, Baltimore, Maryland 21205, USA Department of Medicine, Johns Hopkins University School of Medicine, 855N. Wolfe Street, Rangos 570, Baltimore, Maryland 21205, USA.
References
- 1.Weinhouse S. Isozymes in cancer. Cancer Res. 1971;31:1166–1167. [PubMed] [Google Scholar]
- 2.Shih C, Weinberg RA. Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell. 1982;29:161–169. doi: 10.1016/0092-8674(82)90100-3. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 4.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83:155–158. doi: 10.1007/BF00286709. [DOI] [PubMed] [Google Scholar]
- 6.Sakai T, et al. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Amer J Hum Genet. 1991;48:880–888. [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen KD, et al. Increased methylation variation in epigenetic domains across cancer types. Nature Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boveri T. Concerning the Origin of Malignant Tumors. Williams and Wilkins; 1929. [Google Scholar]
- 9.Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nature Rev Cancer. 2004;4:677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- 10.Lever E, Sheer D. The role of nuclear organization in cancer. J Pathol. 2010;220:114–125. doi: 10.1002/path.2651. [DOI] [PubMed] [Google Scholar]
- 11.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nature Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zullo JM, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149:1474–1487. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 13.Hu S, Cheng L, Wen B. Large chromatin domains in pluripotent and differentiated cells. Acta Biochim Biophys Sin (Shanghai) 2012;44:48–53. doi: 10.1093/abbs/gmr108. [DOI] [PubMed] [Google Scholar]
- 14.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 16.Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nature Rev Cancer. 2012;12:196–209. doi: 10.1038/nrc3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkins RD, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hon GC, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nature Struct Mol Biol. 2011;18:867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nature Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 21.Wen B, et al. Euchromatin islands in large heterochromatin domains are enriched for CTCF binding and differentially DNA-methylated regions. BMC Genomics. 2012;13:566. doi: 10.1186/1471-2164-13-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berman BP, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nature Genet. 2012;44:40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nature Struct Mol Biol. 2011;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nestor C, Ruzov A, Meehan R, Dunican D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques. 2010;48:317–319. doi: 10.2144/000113403. [DOI] [PubMed] [Google Scholar]
- 28.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen ML, et al. Quantification of 5-methylcytosine and 5-hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic-interaction liquid chromatography/quadrupole TOF mass spectrometry. Clin Chem. 2013;59:824–832. doi: 10.1373/clinchem.2012.193938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lian Christine G, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–1146. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang LT, et al. Quantification of the sixth DNA base 5-hydroxymethylcytosine in colorectal cancer tissue and C-26 cell line. Bioanalysis. 2013;5:839–845. doi: 10.4155/bio.13.28. [DOI] [PubMed] [Google Scholar]
- 32.Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 33.Sun Z, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–576. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolf SF, Migeon BR. Studies of X chromosome DNA methylation in normal human cells. Nature. 1982;295:667–671. doi: 10.1038/295667a0. [DOI] [PubMed] [Google Scholar]
- 35.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 36.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228:187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 37.Feinberg AP, Gehrke C. W, Kuo, K. C & Ehrlich, M Reduced genomic 5-methylcytosine content in human colonic neoplasia Cancer Res. 1988;48:1159–1161. [PubMed] [Google Scholar]
- 38.Feinberg AP, Vogelstein B. Hypomethylation of ras oncogenes in primary human cancers. Biochem Biophys Res Commun. 1983;111:47–54. doi: 10.1016/s0006-291x(83)80115-6. [DOI] [PubMed] [Google Scholar]
- 39.De Smet C, et al. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci USA. 1996;93:7149–7153. doi: 10.1073/pnas.93.14.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iacobuzio-Donahue CA, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Amer J Pathol. 2003;162:1151–1162. doi: 10.1016/S0002-9440(10)63911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oshimo Y, et al. Promoter methylation of cyclin D2 gene in gastric carcinoma. Int J Oncol. 2003;23:1663–1670. [PubMed] [Google Scholar]
- 42.Akiyama Y, Maesawa C, Ogasawara S, Terashima M, Masuda T. Cell-type-specific repression of the maspin gene is disrupted frequently by demethylation at the promoter region in gastric intestinal metaplasia and cancer cells. Am J Pathol. 2003;163:1911–1919. doi: 10.1016/S0002-9440(10)63549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho M, et al. Hypomethylation of the MN/CA9 promoter and upregulated MN/CA9 expression in human renal cell carcinoma. Br J Cancer. 2001;85:563–567. doi: 10.1054/bjoc.2001.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura N, Takenaga K. Hypomethylation of the metastasis-associated S100A4 gene correlates with gene activation in human colon adenocarcinoma cell lines. Clin Exper Metastasis. 1998;16:471–479. doi: 10.1023/a:1006589626307. [DOI] [PubMed] [Google Scholar]
- 45.Badal V, et al. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003;77:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Capoa A, et al. DNA demethylation is directly related to tumour progression: evidence in normal, pre-malignant and malignant cells from uterine cervix samples. Oncol Rep. 2003;10:545–549. [PubMed] [Google Scholar]
- 47.Sato N, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63:4158–4166. [PubMed] [Google Scholar]
- 48.Piyathilake CJ, et al. Race- and age-dependent alterations in global methylation of DNA in squamous cell carcinoma of the lung (United States) Cancer Causes Control. 2003;14:37–42. doi: 10.1023/a:1022573630082. [DOI] [PubMed] [Google Scholar]
- 49.Baylin SB, Jones PA. A decade of exploring the cancer epigenome — biological and translational implications. Nature Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones PA, et al. De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci USA. 1990;87:6117–6121. doi: 10.1073/pnas.87.16.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bestor TH. Unanswered questions about the role of promoter methylation in carcinogenesis. Ann NY Acad Sci. 2003;983:22–27. doi: 10.1111/j.1749-6632.2003.tb05959.x. [DOI] [PubMed] [Google Scholar]
- 52.Hosoya K, et al. Adenomatous polyposis coli 1A is likely to be methylated as a passenger in human gastric carcinogenesis. Cancer Lett. 2009;285:182–189. doi: 10.1016/j.canlet.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Levanon D, et al. Absence of Runx3 expression in normal gastrointestinal epithelium calls into question its tumour suppressor function. EMBO Mol Med. 2011;3:593–604. doi: 10.1002/emmm.201100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hitchins MP, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 55.Hitchins MP, Ward RL. Erasure of MLH 1 methylation in spermatozoa-implications for epigenetic inheritance. Nature Genet. 2007;39:1289. doi: 10.1038/ng1107-1289. [DOI] [PubMed] [Google Scholar]
- 56.Bachman KE, et al. Historie modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89–95. doi: 10.1016/s1535-6108(02)00234-9. [DOI] [PubMed] [Google Scholar]
- 57.Sproul D, et al. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc Natl Acad Sci USA. 2011;108:4364–4369. doi: 10.1073/pnas.1013224108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sproul D, et al. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012;13:R84. doi: 10.1186/gb-2012-13-10-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irizarry RA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doi A, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teschendorff A, et al. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4:24. doi: 10.1186/gm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corrada Bravo H, Pihur V, McCall M, Irizarry R, Leek J. Gene expression anti-profiles as a basis for accurate universal cancer signatures. BMC Bioinformatics. 2012;13:272. doi: 10.1186/1471-2105-13-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13:373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol Med. 2007;13:363–372. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 66.Roberts CW, Orkin SH. The SWI/SNF complex-chromatin and cancer. Nature Rev Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 67.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 69.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nature Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 70.Zofall M, et al. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature. 2009;461:419–422. doi: 10.1038/nature08321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young NL, et al. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266–2284. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Voigt P, et al. Asymmetrically modified nucleosomes. Cell. 2012;151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohm JE, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nature Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Widschwendter M, et al. Epigenetic stem cell signature in cancer. Nature Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 77.Chodavarapu RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gupta RA, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu W, et al. Epigenetic silencing of tumour suppressor gene p 15 by its antisense RNA. Nature. 2008;451:202–206. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guil S, Esteller M. Cis-acting noncoding RNAs: friends and foes. Nature Struct Mol Biol. 2012;19:1068–1075. doi: 10.1038/nsmb.2428. [DOI] [PubMed] [Google Scholar]
- 81.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiao Y, et al. DAXX/ATRX, MEN 1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 86.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nature Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 87.Yan XJ, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nature Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 88.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kosmider O, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–1681. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pasqualucci L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nature Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cooper DN, Mort M, Stenson PD, Ball EV, Chuzhanova NA. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum Genomics. 2010;4:406–410. doi: 10.1186/1479-7364-4-6-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 101.Jiang Y, et al. Common fragile sites are characterized by histone hypoacetylation. Hum Mol Genet. 2009;18:4501–4512. doi: 10.1093/hmg/ddp410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fudenberg G, Getz G, Meyerson M, Mirny LA. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature Biotech. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. Cancer genetics of epigenetic genes. Human Mol Genet. 2007;16(Suppl. 1):R28–R49. doi: 10.1093/hmg/ddm021. [DOI] [PubMed] [Google Scholar]
- 104.Varambally S, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 105.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-κB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012;10:678–684. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 107.Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 109.Waddington CH. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology. Allen & Unwin: 1957. [Google Scholar]
- 110.Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 112.Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 113.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci USA. 2010;107(Suppl. 1):1757–1764. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alvarez H, et al. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet. 2011;7:e1001356. doi: 10.1371/journal.pgen.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shah MY. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res. 2010;70:5840–5850. doi: 10.1158/0008-5472.CAN-10-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lande R. Natural selection and random genetic drift in phenotypic evolution. Evolution. 1976;30:314–334. doi: 10.1111/j.1558-5646.1976.tb00911.x. [DOI] [PubMed] [Google Scholar]
- 117.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 118.Kaneda A, Feinberg AP. Loss of imprinting of IGF2: a common epigenetic modifier of intestinal tumor risk. Cancer Res. 2005;65:11236–11240. doi: 10.1158/0008-5472.CAN-05-2959. [DOI] [PubMed] [Google Scholar]
- 119.Timp W, Levchenko A, Feinberg A. P. A new link between epigenetic progenitor lesions in cancer and the dynamics of signal transduction. Cell Cycle. 2009;8:383–390. doi: 10.4161/cc.8.3.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 121.Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol. 1999;32:31–43. doi: 10.1016/s1040-8428(99)00019-0. [DOI] [PubMed] [Google Scholar]
- 122.Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cui H, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- 124.Sakatani T, et al. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science. 2005;307:1976–1978. doi: 10.1126/science.1108080. [DOI] [PubMed] [Google Scholar]
- 125.Teschendorff AE, Widschwendter M. Differential variability improves the identification of cancer risk markers in DNA methylation studies profiling precursor cancer lesions. Bioinformatics. 2012;28:1487–1494. doi: 10.1093/bioinformatics/bts170. [DOI] [PubMed] [Google Scholar]
- 126.Siddique H, Zou JP, Rao VN, Reddy E. The BRCA2 is a histone acetyltransferase. Oncogene. 1998;16:2283. doi: 10.1038/sj.onc.1202003. [DOI] [PubMed] [Google Scholar]
- 127.Fuks F, Milner J, Kouzarides T. BRCA2 associates with acetyltransferase activity when bound to P/CAF. Oncogene. 1998;17:2531. doi: 10.1038/sj.onc.1202475. [DOI] [PubMed] [Google Scholar]
- 128.Esteve PO, Chin HC, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci USA. 2005;102:1000–1005. doi: 10.1073/pnas.0407729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu P, et al. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 130.Campbell PM, Szyf M. Human DNA methyltransferase gene DNMT1 is regulated by the APC pathway. Carcinogenesis. 2003;24:17–24. doi: 10.1093/carcin/24.1.17. [DOI] [PubMed] [Google Scholar]
- 131.Sun L, et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal. 2007;19:2255–2263. doi: 10.1016/j.cellsig.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 132.Lofton-Day C, et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin Chem. 2008;54:414–423. doi: 10.1373/clinchem.2007.095992. [DOI] [PubMed] [Google Scholar]
- 133.Warren JD, et al. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011;9:133. doi: 10.1186/1741-7015-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zou H, et al. Highly methylated genes in colorectal neoplasia: implications for screening. Cancer Epidemiol Biomarkers Prev. 2007;16:2686–2696. doi: 10.1158/1055-9965.EPI-07-0518. [DOI] [PubMed] [Google Scholar]
- 135.Lee WH, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA. 1994;91:11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhuang J, et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012;8:el002517. doi: 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li M, et al. Sensitive digital quantification of DNA methylation in clinical samples. Nature Biotech. 2009;27:858–863. doi: 10.1038/nbt.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Silber JR, Bobola MS, Blank A, Chamberlain MC. O6-Methylguanine-DNA methyltransferase in glioma therapy: promise and problems. Biochim Biophys Acta. 2012;1826:71–82. doi: 10.1016/j.bbcan.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nature Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 140.Knutson SK, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nature Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 141.Fiskus W, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Popovici-Muller J, et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HC in vivo. ACS Med Chem Lett. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-mol ecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hull MA. Nutritional agents with anti-inflammatory properties in chemoprevention of colorectal neoplasia. Recent Results Cancer Res. 2013;191:143–156. doi: 10.1007/978-3-642-30331-9_8. [DOI] [PubMed] [Google Scholar]
- 145.Kraus S, Naumov I, Arber N. COX-2 active agents in the chemoprevention of colorectal cancer. Recent Results Cancer Res. 2013;191:95–103. doi: 10.1007/978-3-642-30331-9_5. [DOI] [PubMed] [Google Scholar]
- 146.Joshi PH, et al. A point-by-point response to recent arguments against the use of statins in primary prevention: this statement is endorsed by the American Society for Preventive Cardiology. Clin Cardiol. 2012;35:404–409. doi: 10.1002/clc.22016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yuasa Y, et al. Insulin-like growth factor 2 hypomethylation of blood leukocyte DNA is associated with gastric cancer risk. Int J Cancer. 2012;131:2596–2603. doi: 10.1002/ijc.27554. [DOI] [PubMed] [Google Scholar]
- 148.Kaneda A, et al. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc Natl Acad Sci USA. 2007;104:20926–20931. doi: 10.1073/pnas.0710359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 150.Morin RD. et ai. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kanai Y, Ushijima S, Nakanishi Y, Sakamoto M, Hirohashi S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003;192:75–82. doi: 10.1016/s0304-3835(02)00689-4. [DOI] [PubMed] [Google Scholar]
- 152.Tefferi A, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905–911. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Langemeijer SM, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nature Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 154.Yan H, et al. IDH1 and IDH2 mutations in gliomas. New Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gui Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ward R, Johnson M, Shridhar V, van Deursen J, Couch FJ. CBP truncating mutations in ovarian cancer. J Med Genet. 2005;42:514–518. doi: 10.1136/jmg.2004.025080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 159.Kishimoto M, et al. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res. 2005;11:512–519. [PubMed] [Google Scholar]
- 160.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cromer MK, et al. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97:E1774–E1781. doi: 10.1210/jc.2012-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pugh TJ et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ernst T, et al. Inactivating mutations of the histone methyltransferase gene EZM2 in myeloid disorders. Nature Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 167.Bodor C, et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia. 2011;25:726–729. doi: 10.1038/leu.2010.311. [DOI] [PubMed] [Google Scholar]
- 168.Guichard C, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Network TCCA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Biegel JA, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- 174.Woodson K, et al. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J Natl Cancer Inst. 2004;96:407–410. doi: 10.1093/jnci/djh042. [DOI] [PubMed] [Google Scholar]
- 175.Yun K, Soejima H, Merrie AEH, McCall JL, Reeve AE. Analysis of IGF2 gene imprinting in breast and colorectal cancer by allele specific-PCR. J Pathol. 1999;187:518–522. doi: 10.1002/(SICI)1096-9896(199904)187:5<518::AID-PATH276>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 176.Nakagawa M, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 177.Choi JH, et al. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Halkidou K, et al. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 179.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 180.Lin Z, et al. Combination of proteasome and HDAC inhibitors for uterine cervical cancer treatment. Clin Cancer Res. 2009;15:570–577. doi: 10.1158/1078-0432.CCR-08-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kleer CG, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ozdag H, et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 2006;7:90. doi: 10.1186/1471-2164-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Huang BH, et al. Inhibition of histone deacetylase 2 increases apoptosis and p21 Cipl/WAFl expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- 184.Wilson AJ, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Z, et al. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10:6962–6968. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- 186.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. 2009;284:3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ashraf N, et al. Altered sirtuin expression is associated with node-positive breast cancer. Br J Cancer. 2006;95:1056–1061. doi: 10.1038/sj.bjc.6603384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lu PJ, et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–15645. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- 189.Silva FP, et al. Enhanced methyltransferase activity of SMYD3 by the cleavage of its N-terminal region in human cancer cells. Oncogene. 2008;27:2686–2692. doi: 10.1038/sj.onc.1210929. [DOI] [PubMed] [Google Scholar]
- 190.Northcott PA, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Peng DF, et al. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis. 2006;27:1160–1168. doi: 10.1093/carcin/bgi361. [DOI] [PubMed] [Google Scholar]
- 192.Saito Y, et al. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer. 2003;105:527–532. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- 193.Nakagawa T, et al. DNA hypermethylation on multiple CpG islands associated with increased DNA methyltransferase DNMT1 protein expression during multistage urothelial carcinogenesis. J Urol. 2005;173:1767–1771. doi: 10.1097/01.ju.0000154632.11824.4d. [DOI] [PubMed] [Google Scholar]
- 194.Agoston AT, et al. Increased protein stability causes DNA methyltransferase 1 dysregulation in breast cancer. J Biol Chem. 2005;280:18302–18310. doi: 10.1074/jbc.M501675200. [DOI] [PubMed] [Google Scholar]
- 195.Butcher DT, Rodenhiser DI. Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours. Eur J Cancer. 2007;43:210–219. doi: 10.1016/j.ejca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 196.McCarthy H, et al. High expression of activation-induced cytidine deaminase (AID) and splice variants is a distinctive feature of poor-prognosis chronic lymphocytic leukemia. Blood. 2003;101:4903–4908. doi: 10.1182/blood-2002-09-2906. [DOI] [PubMed] [Google Scholar]