

Abstract
Chromosomal instability (CIN) is a hallmark of solid tumor biology and is implicated in carcinogenesis. Preferentially eliminating malignant cells by targeting CIN and aneuploidy is an attractive anti-neoplastic strategy. We previously reported that CDK2 antagonism causes lung cancer cells to undergo anaphase catastrophe and apoptosis through inhibition of phosphorylation of the centrosomal protein CP110. Cells with activating KRAS mutations were particularly sensitive to CDK2 inhibition due to down-regulation of CP110 protein levels. This study investigated mechanisms of CDK2 antagonism that mediate anaphase catastrophe via changes in CP110 protein expression and how activated KRAS affects CP110 levels in lung cancers. Site-direct mutagenesis revealed candidate CDK phosphorylation sites of CP110 (residues Ser 170 and Thr 194) critical for conferring anaphase catastrophe by altering centrosome clustering in mitosis. Intriguingly, KRAS mutation can promote CP110 protein degradation by upregulating the ubiquitin ligase SCFcyclinF, which targets CP110 protein for destabilization. Finally, CDK2 inhibitor response was enhanced when combined with knockdown of the deubiquitinase USP33 that in turn accelerates CP110 protein degradation. Thus, this study provides molecular pharmacologic insights into how CP110 expression regulates response to CDK2 inhibition. An improved understanding of in vitro antineoplastic mechanisms of combining CDK2 antagonism with induced CP110 repression provides a rationale for exploring clinical consequences of this strategy. Taken together, preclinical findings obtained from combining CDK2 inhibition with USP33 repression have implications for treating patients with non-small cell lung cancers.
Keywords: CP110, anaphase catastrophe, centrosome clustering, KRAS mutation and lung cancer
Introduction
Chromosomal instability (CIN) is a hallmark of solid tumor biology and is one feature that differentiates normal and malignant cells (1). CIN is a driver of tumor initiation and growth since it facilitates tumor adaptation (2,3). A direct consequence of CIN is aneuploidy, which has long been studied for its effect on tumorigenesis and correlation with aggressiveness and tumor stage (1).
While gain of CIN can accelerate tumorigenesis, excessive instability is physiologically intolerable and cytotoxic (4–6). Therapeutic targeting of CIN and aneuploidy have been proposed by either suppressing or critically elevating the CIN rate (1). Strategies that target CIN include regulating proteins or pathways that are essential for CIN, but dispensable for normal cells. Tumor cells rely on several pathways to tolerate aneuploidy including the ubiquitin-proteasome, autophagy, metabolic stress response and energy stress inducing pathways (7). Several CIN targets are undergoing preclinical testing, including Aurora B kinase (7–12), HSET (7,13,14), heat shock protein (HSP) chaperone, autophagy and the ubiquitin-proteasome (15–18), but none have yet been FDA-approved. Alternative methods of targeting CIN should be investigated.
Our prior lung cancer work found that CDK2 inhibition causes anaphase catastrophe, a lethal event where cells with supernumerary centrosomes segregate chromosomes into more than two daughter cells, resulting in non-viable daughter cells (19). CP110, a centrosomal protein, was uncovered as a mediator of CDK2-inhibitor-induced anaphase catastrophe (20). Furthermore, KRAS mutations sensitized lung cancers to CDK2-inhibitor-mediated anaphase catastrophe by down-regulating CP110 levels (20). This study further investigates the CP110 role in anaphase catastrophe.
CP110 is a centrosomal protein that critically regulates centrosome duplication and separation, chromosome segregation and cilia formation (21–33). It is a direct substrate of cyclin E-CDK2, cyclin A-CDK2, and cyclin B-CDK1 (21). CP110 has ten putative CDK phosphorylation sites and CP110 with eight of these CDK phosphorylation sites mutated (CP110-MUT) conferred polyploidy (21). Notably, CP110-MUT was no longer a substrate for CDKs as confirmed by kinase assays although centrosomal localization of CP110-MUT was unaffected (21). Low CP110 protein level or loss of CP110 phosphorylation promoted unscheduled centrosome separation (21). Roles of individual CP110-CDK phosphorylation sites in anaphase catastrophe were not previously determined.
CP110 protein levels are tightly regulated through the cell cycle. Levels are low in quiescent cells (G0 phase) or early G1 phase, but markedly increase as cells progress through the G1-S transition. CP110 levels start to decline in the G2 cell cycle phase and diminish after cells complete mitosis (21). CP110 levels are controlled transcriptionally, post-translationally and by miRNAs (21–28) to prevent centriole duplication, cytokinesis and primary cilia formation errors (21,22,29–32). In G2, CP110 complexes with the F-box protein cyclinF and is ubiquitinated by the SCFcyclinF E3 ubiquitin ligase complex and degraded. The siRNA-mediated depletion of cyclin F in G2 induces centrosomal and mitotic abnormalities, which can be reverted by co-silencing CP110 (22). SCFcyclinF mediated degradation of CP110 is required for fidelity of mitosis and genomic integrity (22).
CP110 deubiquitinases also regulate CP110 protein levels (22). Both USP33 and USP20 are deubiquitinases that recognize CP110 and counteract SCFcyclinF ubiquitin ligase activity. USP33 was determined to exert marked effects on CP110 protein expression (22). USP33 depletion did not inhibit centriole duplication, but inhibited centrosome re-duplication, which is similar to the effect of CP110 depletion (24).
Since centrosome amplification is associated with tumorigenesis in multiple human cancers and USP33 directly affects centrosome duplication by regulating CP110 protein expression, the role of USP33 in cancer biology is under active investigation. This study assesses the relationship between KRAS mutation and CP110 degradation by titrating USP33 levels.
The current study tested the hypothesis that lung cancer cells were sensitized to CDK2-inhibitor-mediated anaphase catastrophe by targeting for repression the CP110 deubiquitinase, USP33. The obtained findings provide a mechanistic understanding of CP110 CDK phosphorylation site functions and also the relationship between KRAS mutation and a specific CP110 protein degradation pathway. Therapeutic intervention is proposed where antagonism of CDK2 along with engaging a specific CP110 degradation pathway would exert substantial antitumor activity in aneuploid lung cancers.
Materials and Methods
Chemicals and antibodies
Seliciclib (CYC202, R-roscovitine) was provided by Cyclacel, Ltd (10mM stock solution in dimethyl sulfoxide, DMSO). The seliciclib dosage used (10μM) is clinically achievable (34) and biological effects of seliciclib at 10μM were due to CDK2 inhibition rather than CDK7/9 blockade in lung cancer cells (19). Antibodies used were: α-tubulin (T6199, Sigma Aldrich, (1:1000 immunofluorescence and 1:10000 immunoblot), α-tubulin (YL1/2) (NB600-506, Novus Biologicals, 1:1500), γ-tubulin (T5326, Sigma Aldrich, 1:1000), CP110 (12780-1-AP, Proteintech, 1:750), HA.11 clone 16B12 monoclonal antibody (MMS-101P, Covance, 1:3000), cyclin F (C-20) (SC-952, Santa Cruz Biotechnology, 1:500), CEP76 (A302-326A, Bethyl Laboratories, 1:1000), USP33 (A300-925A, Bethyl Laboratories, 1:1000 in 5%BSA) and KRAS (ab55391, Abcam, 1:1000). Secondary antibodies used were: Texas red anti-mouse IgG (H+L) (TI-2000, Vector Laboratories.), Fluorescein anti-mouse IgG (H+L) (FI-2000, Vector Laboratories.), Alexa fluor 594 donkey anti-rat IgG (H+L) (A21209, Invitrogen), ECL anti-rabbit lgG (NA934V, GE Healthcare), ECL anti-mouse lgG (NA931V, GE Healthcare) and horseradish peroxidase–conjugated donkey anti-goat IgG (sc-2020, Santa Cruz Biotechnology.). Hoechst 33342 (62249, Thermo Scientific, 1:25000) stained DNA. Pro-Long Gold anti-fade reagent (P36934, Invitrogen) preserved immunofluorescence.
Cell culture
Human lung cancer cell lines A549, H1703, SW900, H2122, H460 and H522 were purchased from American Type Culture Collection (ATCC) between 1998 and 2012. HOP62 cells were purchased from the National Cancer Institute in 2012. These different cell lines were not independently authenticated. Murine lung cancer cell lines LKR13, 344P and 393P were kindly provided by Dr. Johnathan Kurie, (MD Anderson Cancer Center) between 2014 and 2015 and were authenticated by immunoblot analyses expression of the engineered KRAS species (20). ED-1 murine lung cancer cell line were developed in our laboratory (Geisel School of Medicine at Dartmouth) in 2009. Frozen stocks of this original cell line were prepared and used in this study after authentication (35).
Expression plasmids and transient transfection
Expression vectors HA-tagged wild-type pcDEF3-CP110 (CP110-WT) and CP110 with 8 phosphorylation sites mutated, pcDEF3-CP110 (CP110-MUT) were previously reported (21). Logarithmically growing ED-1, LKR13, Hop62 and H522 cell lines were each transiently transfected using Lipofectamine 2000 reagent (Invitrogen). Each experiment was independently replicated at least three times.
Indicated lung cancer cells were transfected with small interfering RNAs (siRNAs) using Lipofectamine 2000 (Invitrogen). The siRNA-mediated silencing was confirmed by immunoblot analyses. The siRNA sequences appear in Supplemental Table S1. Each experiment was independently replicated at least three times.
Site-directed mutagenesis
To generate specific mutations of CP110-MUT where eight potential serine and threonine phosphorylation sites were mutated to alanine residues (Ser 45, Ser 170, Thr 194, Ser 366, Ser 372, Ser 400, Ser 516 and Thr 566), each site was replaced by a serine or threonine residue to restore wild-type sequences at individual or multiple sites using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). To confirm effects of specific phosphorylation sites on anaphase catastrophe and centrosome clustering, Ser 170, Thr 194 of CP110-WT were replaced by alanine residues individually and independently at both sites.
Multipolar anaphase assay
Following treatments of the indicated lung cancer cells, cells were fixed in cold methanol, incubated with the anti-α-tubulin–specific antibody and independently mounted with Pro-Long Gold anti-fade reagent (Invitrogen). Stained cells were examined using an Eclipse TE 2000-E microscope (Nikon). Anaphase cells that contained three or more spindle poles were scored as multipolar. Data were expressed as percent multipolar versus total anaphase cells.
Effective centrosome clustering analysis assay
The indicated lung cancer cells were fixed in cold methanol, incubated with anti-α-tubulin–specific antibody and anti-γ-tubulin-specific antibody, and independently mounted with Pro-Long Gold anti-fade reagent (Invitrogen). Stained cells were examined using an Eclipse TE 2000-E microscope (Nikon). Anaphase cells that contained two spindle poles, but had more than one centrosome at either pole were scored as effective bipolar centrosome clustering. Data were expressed as percent effective centrosome clustering versus total anaphase cells.
Generation of stable KRAS transfectants
Logarithmically growing ED-1 (3 × 106) and H522 (5 × 106) cells were individually plated onto 10 cm tissue culture dishes 24 hours before transfection. Twelve μg each of the pCGN K-RasG12V, 188L plasmid (Addgene) with the pPUR expression plasmid (Clontech) or an empty vector with the pPUR plasmid were individually transfected into ED-1 or H522 cells using Lipofectamine 2000 (invitrogen) and Opti-MEM medium (Gibco). Puromycin selection began 24 hours after transfection. Engineered KRAS overexpression was confirmed by immunoblot analysis.
CP110 protein stability assay
To assess CP110 protein stability in cells with or without KRAS mutation, respectively, cells were transfected with CP110-WT expressing vectors 24 hours before treatment with or without cycloheximide (CHX, 40μg/ml, Sigma) for indicated time periods. Cells were collected and processed for immunoblot analysis.
Proliferation assay
Logarithmically growing cells were plated onto individual 12-well tissue culture plates (8 × 103 cells/well). Twenty-four hours later, cells were transfected with siRNAs targeting USP33 or control siRNAs. Twenty-four hours after transfection, cells were treated with 10μM seliciclib versus vehicle controls. Three independent wells were seeded in each experiment with triplicate independent replicates. Proliferation was measured using the CellTiter-Glo Assay Kit (Promega), as described (37). Trypan blue viability assays were performed (35).
Immunoblot analyses
Cells were lysed with ice-cold radioimmunoprecipitation (RIPA) buffer supplemented with protease inhibitors (Biosciences), and immunoblot analyses were done, as before (38). Lysates were size-fractionated using previously optimized methods (38) by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Schleicher and Schuell Bioscience) and incubated with the indicated antibodies.
Statistical analyses
Results of independent experiments were pooled to assess statistical significance. Two-tailed t-tests were used. Statistical significance was noted with these symbols: *, P < 0.05 and **, P < 0.01.
Results
Mutation of CDK phosphorylation sites of CP110 and anaphase catastrophe
To determine the role of potential CDK phosphorylation sites of CP110 in CDK2 inhibitor-mediated anaphase catastrophe, a CP110 expression vector was generated with all ten putative CDK phosphorylation sites replaced by alanine residues (CP110-ALL MUT). While CP110-WT protected cells from CDK2 inhibitor-mediated anaphase catastrophe in lung cancer cells, neither CP110-MUT nor CP110-ALL MUT did. (Supplemental Fig. S1).
To determine which CDK phosphorylation site(s) is/are critical in mediating anaphase catastrophe, each of the eight potential phosphorylation-sites in CP110-MUT were restored to the wild-type sequence with serine or threonine residues, respectively. ED-1 cells were independently transfected with the indicated CP110 mutants, CP110-WT or an insertless vector, and then treated with seliciclib. Anaphase catastrophe was quantified 24 hours after these treatments. Restoration of the Ser 170 and Thr 194 sites, but not of other CP110 sites significantly recovered the ability of CP110 to prevent anaphase catastrophe from being induced by seliciclib treatment as compared to transfection of the CP110-MUT expression vector in murine (Fig. 1A) or in human (Supplemental Fig. S2A) lung cancer cells. Combined restoration of Ser 170 and Thr 194 from the alanine residues of the CP110-MUT vector to wild-type sequences restored the ability of CP110 to reduce anaphase catastrophe after seliciclib treatment (Fig. 1B and Supplemental Fig. S2A, structure seen in Fig. 1D).
Fig. 1.
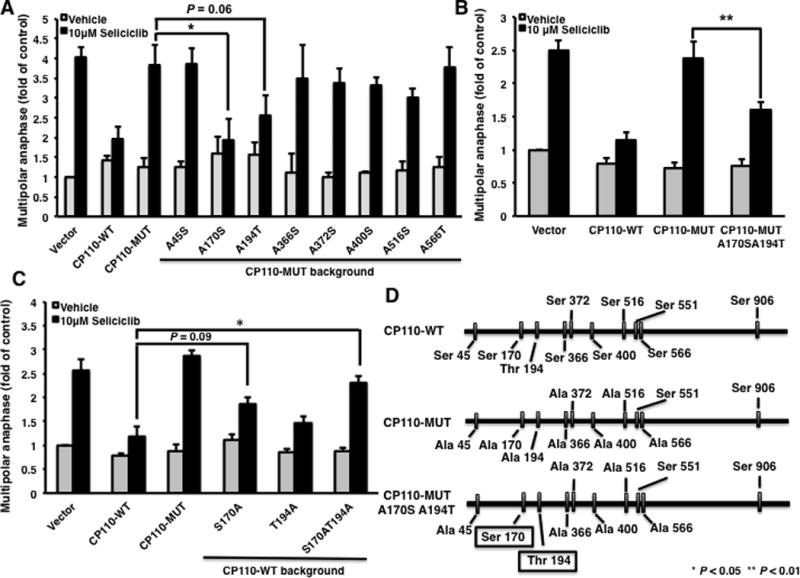
Effects of individual CP110 phosphorylation sites on Cdk2-inhibition-mediated anaphase catastrophe in murine lung cancer cells. (A) ED-1 cells were transfected with the CP110-MUT expressing vector with the indicated phosphorylation sites restored to wild-type sequences. Twenty-four hours after transfection, cells were treated with seliciclib (10μM) for 24 hours and fixed and scored for multipolar anaphases. (B) Transfection effects of CP110-MUT with both Ser 170 and Thr 194 restored to wild-type sequences on CDK2-inhibition-mediated anaphase catastrophe in ED-1 cells. (C) Transfection effects of CP110-WT expressing vector having Ser 170 and Thr 194 individually or together replaced with alanine residues on CDK2-inhibition-mediated anaphase catastrophe in ED-1 cells. (D) Schematic of CP110-WT, CP110-MUT (8 sites) and CP110-MUT with Ser 170 and Thr 194 restored to wild-type sequences. All experiments were independently replicated at least three times.
To confirm the effects of modification of residues Ser 170 and Thr 194 of CP110 in mediating anaphase catastrophe, Ser 170 and Thr 194 were individually or both substituted for an alanine residue within the CP110-WT expression vector. Inactivation of Ser 170 somewhat impaired how CP110 antagonized anaphase catastrophe after treatment with the CDK2 inhibitor seliciclib, while inactivation of both sites disrupted the ability of CP110 to antagonize anaphase catastrophe formation in murine (Fig. 1C) and human (Supplemental Fig. S2B) lung cancer cells. Taken together, candidate CDK phosphorylation sites of CP110 Ser 170 and Thr 194 were found to be critical for protecting cells from anaphase catastrophe caused by CDK2 inhibition.
CDK phosphorylation of CP110 regulates centrosome clustering
CP110 regulates centrosome maturation and separation during mitosis (20–22,24,27,29,30). Our prior work established that anaphase catastrophe occurs after CDK2 inhibition and that CP110 CDK phosphorylation sites are critical for these functions (20). Antagonism of anaphase catastrophe is achieved by inhibiting centrosome overduplication or by clustering supernumerary centrosomes to form bipolar spindles during mitosis. It was hypothesized that CP110 would decrease anaphase catastrophe by clustering extra centrosomes to form bipolar spindles during mitosis and that specific CDK phosphorylation sites are critical for centrosome clustering. Effective centrosome clustering was scored as cells undergo bipolar anaphase with more than one centrosome on either or both ends of the spindle (Fig. 2A).
Fig. 2.
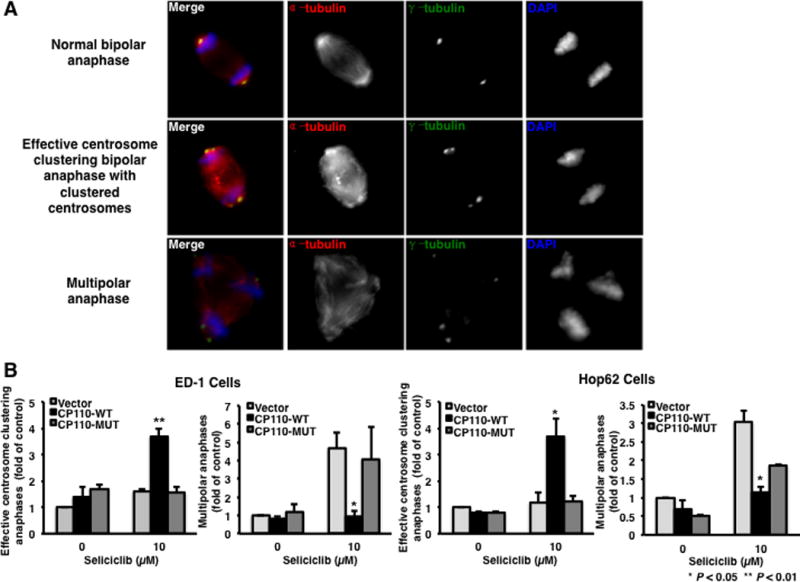
Effects of different candidate CP110 phosphorylation sites on centrosome clustering when independently challenged with a CDK2 inhibitor in murine and human lung cancer cells. (A) Representative examples of normal bipolar anaphase with one centrosome at each end of spindles (upper panel); anaphase with supernumerary centrosomes clustered to form bipolar spindles (middle panel) and anaphase with multiple centrosomes and that also form multipolar spindles (bottom panel) in ED-1 murine lung cancer cells. (B) ED-1 and Hop62 cells were each transfected with an empty vector, a CP110-WT or a CP110-MUT expressing plasmid. Twenty-four hours after transfection, cells were treated with seliciclib (10μM) for 24 hours and fixed and scored for effective centrosome clustering and multipolar anaphases. All experiments were independently replicated at least three times.
To investigate whether CP110 directly affects centrosome clustering in lung cancer cells, ED-1 and Hop62 cells were independently transfected with CP110-WT, CP110-MUT or an insertless expression vector, and treated with seliciclib for 24 hours before quantification of centrosome clustering and anaphase catastrophe. Expression of CP110-WT, but not of a CP110-MUT expression vector significantly increased effective centrosome clustering and decreased anaphase catastrophe rates after CDK2 inhibition in both murine (P < 0.01) and human (P < 0.05) lung cancer cells (Fig. 2B).
Since residues Ser 170 and Thr 194 proved critical for regulating anaphase catastrophe, their potential roles in regulating centrosome clustering were next investigated. ED-1 and Hop62 cells were independently transfected with the indicated CP110 mutant, CP110-WT or an insertless expression vector, and treated with seliciclib. Effective centrosome clustering was quantified 24 hours later. Restoration of Ser 170 or Thr 194 or both residues to wild-type sequences, but not two randomly chosen CDK phosphorylation sites of the CP110-MUT expression vector, significantly recovered the function of CP110 in promoting effective centrosome clustering when treated with seliciclib treatment (P < 0.05) (Fig. 3A and B, left panel). Consistently, inactivation of Ser 170 or Thr 194 or both sites of the CP110-WT expression vector significantly decreased effective centrosome clustering occurrence rates after CDK2 inhibition (P < 0.05) (Fig. 3A and B, right panel).
Fig 3.
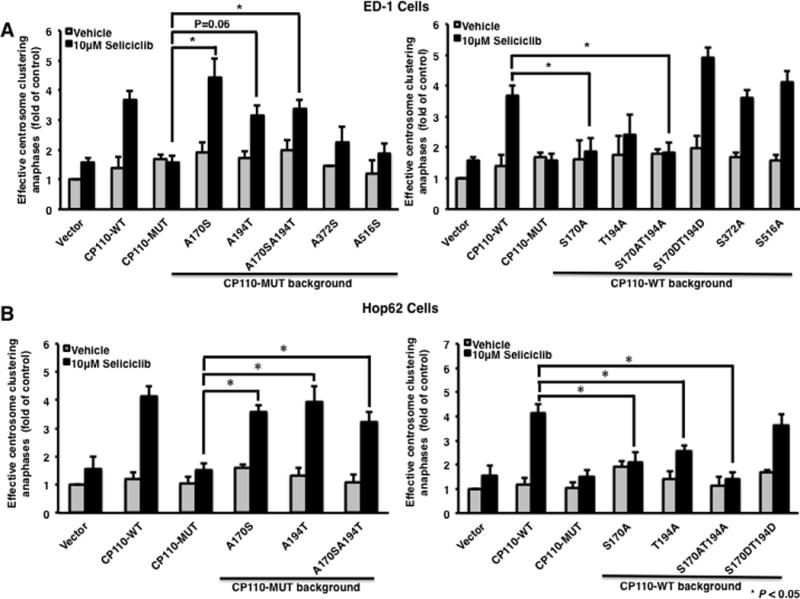
Effects of Ser 170 and Thr 194 CP110 phosphorylation sites on centrosome clustering when independently challenged with a CDK2 inhibitor in murine and human lung cancer cells. (A) ED-1 and (B) Hop62 cells were transfected with (left panel) the CP110-MUT expression vector with indicated phosphorylation sites restored to wild-type residues or (right panel) the CP110-WT expression vector or with a vector having the indicated phosphorylation sites replaced with alanine or aspartic acid. Twenty-four hours after transfection, cells were treated with seliciclib (10μM) for 24 hours and fixed and scored for effective centrosome clustering. All experiments were independently replicated at least three times.
Thus, CP110 controlled anaphase catastrophe caused by CDK2 inhibition in lung cancer cells by promoting effective centrosome clustering. Also, candidate CP110 CDK phosphorylation sites and specifically residues Ser 170 and Thr 194 were involved in conferring this regulation.
KRAS decreases CP110 protein expression by promoting its degradation
KRAS mutation sensitized lung cancer cells to anaphase catastrophe by decreasing CP110 protein, but not mRNA expression levels, indicating that this down-regulation was post-transcriptional (20). Yet, the level of this post-transcriptional regulation was not determined. To investigate whether KRAS mutation directly affects CP110 degradation, KRAS or an insertless expression vector was each stably and individually transfected into ED-1 (KRAS-ED-1 and EV-ED-1) and H522 (KRAS-H522 and EV-H522) cells. Cells were then independently transfected with the CP110-WT expression vector. Degradation rates were determined using a cycloheximide (CHX) treatment time course to inhibit de novo protein synthesis 24 hours after transfection. The CP110-WT species was degraded more rapidly in KRAS-ED-1 and KRAS-H522 cells as compared with EV-ED-1 and EV-H522 transfectants (Fig. 4A and 4B).
Fig. 4.
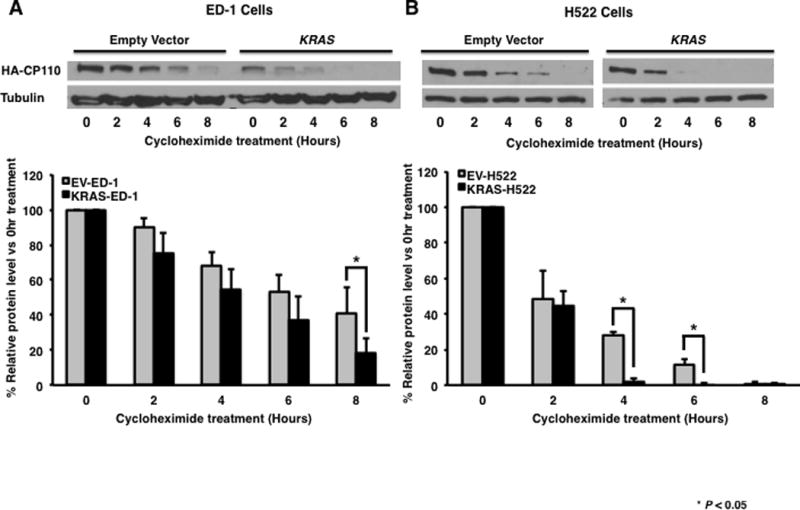
CP110 protein stability assays done independently in human (H522) and murine (ED-1) lung cancer cells that respectively had or not a KRAS mutation introduced. Empty vector or KRAS stably transfected (A) ED-1 and (B) H522 cells were transfected with the CP110-WT expression vector. Twenty-four hours later, cells were treated with or without cycloheximide (CHX, 40μg/ml) for the indicated time periods. Expression of CP110 protein at each time point was shown using immunoblot analyses and ImageJ quantification. Experiments were independently replicated at least three times. Quantifications by ImageJ were achieved by pooling independent experimental results.
To investigate whether CP110 phosphorylation affected the CP110 degradation rate, CP110-WT or CP110-MUT was independently transfected into KRAS wild-type ED-1 cells. The degradation rates were determined with or without cycloheximide (CHX) treatment 24 hours after transfection. There was no appreciable difference between the half-life of CP110-WT and CP110-MUT in ED-1 cells (Supplemental Fig. S3). Thus, KRAS mutation accelerates the CP110 degradation rate in lung cancer cells and this is independent of CP110 phosphorylation status.
KRAS decreases CP110 levels by regulating its degradation machinery
To explore mechanisms responsible for activated-KRAS-conferred CP110 degradation, proteins involved in CP110 degradation were investigated in murine (Fig. 5A) lung cancer cells. The basal level of cyclin F, a key player in CP110 level regulation (22), is lower in KRAS wild-type murine lung cancer cells as compared to KRAS mutant ones (Fig. 5A). The basal levels of USP33 and CEP76, two proteins that regulate CP110 expression (24,33), did not show a trend in KRAS wild-type versus KRAS mutant murine lung cancer cells (Fig. 5A). Cyclin F basal levels were also upregulated in KRAS-ED-1 cells as compared to EV-ED-1 cells (Fig. 5B).
Fig. 5.
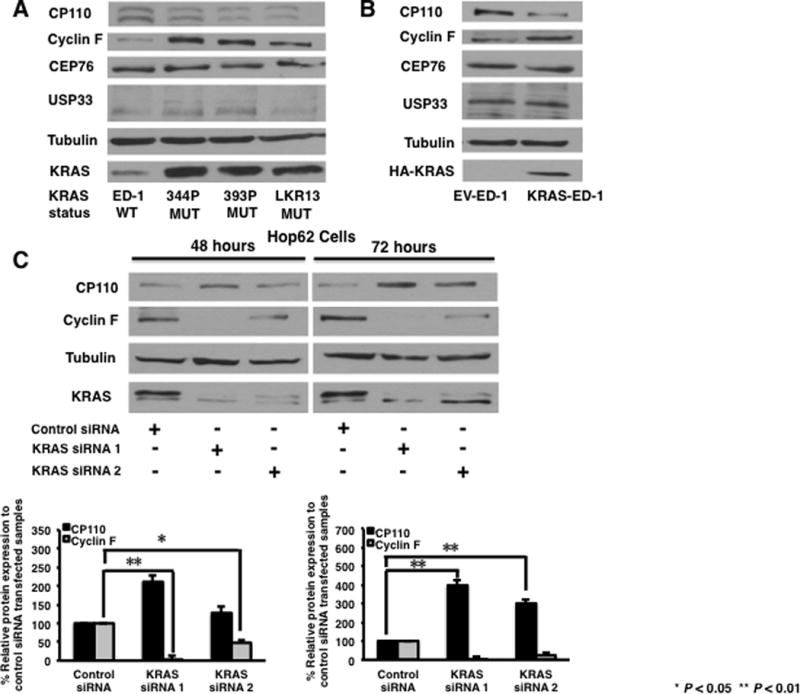
Effects of KRAS on expression of cyclin F and CP110 in human and murine lung cancer cells. (A) Respective CP110, cyclin F, CEP76, USP33 and RAS protein levels in murine lung cancer cell lines. (B) Respective CP110, cyclin F, CEP76, USP33 and RAS protein levels in empty vector or KRAS expression vector stably transfected ED-1 cells. (C) LKR13 and Hop62 cells were independently transfected with each of two different KRAS targeting siRNAs or control siRNA. Effects of KRAS knock-down at 48 and 72 hours on CP110 and cyclin F expression. Expression of CP110 and cyclin F protein at each time point was shown using immunoblot analyses and ImageJ quantification. Experiments were independently replicated at least three times. Quantification by ImageJ was achieved by pooling independent experimental results.
To confirm and extend analyses of the role of KRAS in regulating expression of cyclin F, transient KRAS knockdown was achieved in LKR13 and Hop62 cells using siRNAs. Decreased KRAS expression was detected at 48 and 72 hours after transfection (Fig. 5C). Decreased cyclin F expression was detected at both 48 and 72 hours after transfection and increased CP110 expression was evident 72 hours after transfection in both cell lines (Fig. 5C). This order of changes of KRAS, cyclin F and CP110 expression profiles implied that KRAS down-regulates CP110 level by up-regulating cyclin F.
Combining CDK2 antagonism and USP33 depletion augments anaphase catastrophe
Since both CDK2 inhibition and KRAS mutation affect CP110 pathways and anaphase catastrophe, it was determined if lung cancer cells are sensitized to CDK2 inhibition by combining this with induced repression of CP110 levels. It was hypothesized that dual targeting of these pathways would augment anaphase catastrophe. USP33 is a deubiquitinating enzyme of CP110 that can antagonize SCFcyclinF-mediated CP110 ubiquitination and stabilize CP110 (24). To discern effects of USP33 knockdown along with CDK2 inhibition on anaphase catastrophe in lung cancer cells, two different siRNAs targeting murine Usp33 and human USP33 and a scrambled control siRNA were each used. Marked knockdown of USP33 was achieved in murine and human cells as validated by immunoblot analyses (Fig. 6 and Supplemental Fig. S4). As expected, after 48 hours of USP33 knockdown, reduced levels of CP110 were achieved in A549 and Hop62 human lung cancer cells (Fig. 6A and 6B). Knock-down of Usp33 did not affect CP110 levels in ED-1 murine lung cancer cells (Supplemental Fig.S4). A549 and Hop62 were treated with seliciclib for 24 hours after USP33 knockdown before anaphase catastrophe analyses. Knockdown of USP33 significantly (P <0.01) augmented anaphase catastrophe in A549 and Hop62 cells. When combined with CDK2 inhibition, USP33 knockdown increased anaphase catastrophe caused by seliciclib treatment in both lung cancer cell lines (Fig. 6A and 6B). USP33 knock-down increased growth inhibition conferred by seliciclib treatment in human lung cancer cell lines (Supplemental Fig. S5).
Fig. 6.
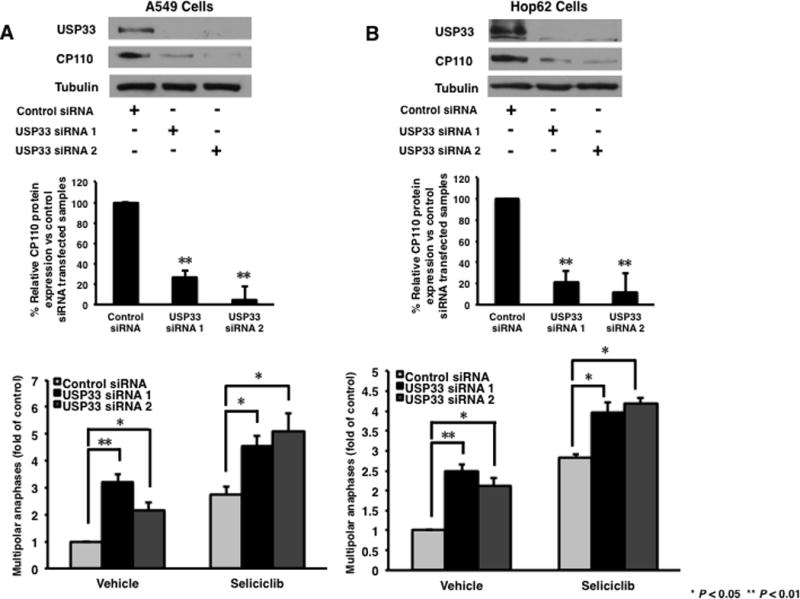
Effects of USP33 knock-down on CP110 protein levels and anaphase catastrophe in human lung cancer cells. (A) A549 and (B) Hop62 cells were independently transfected with each of two different USP33 targeting siRNAs or control siRNA. Forty-eight hours after USP33 knockdown, cells were treated with seliciclib (10μM) for 24 hours and fixed and scored for multipolar anaphases. Expression of CP110 protein at each time point was shown using immunoblot analyses and ImageJ quantification. Experiments were independently replicated at least three times. Quantifications by ImageJ were done by combining for analyses all independent experimental results.
Discussion
Prior work revealed that CDK2 antagonism caused multipolar cell division and this led to anaphase catastrophe in lung cancers (19). Also CP110, a direct target of cyclin E-CDK2, was found as a mediator of this effect (20). That prior work was built upon here by showing specific CP110 CDK phosphorylation sites were involved in anaphase catastrophe. Specific sites promoted centrosome clustering in lung cancer cells and thereby protected these cells from undergoing anaphase catastrophe. CP110 is not known to have enzymatic activity, but serves a structural role in regulating centrosome duplication and separation, chromosome segregation and cilia formation by interacting with several distinct protein complexes (21,22,24,27–33). Residues Ser 170 and Thr 194 of CP110 are located between the coiled-coil and destruction box (D-box) domains (21). It is likely that these two sites are required for interactions between CP110 and by this other centrosomal proteins that regulate centrosome clustering.
Centrosomes represent an attractive target for anti-cancer therapy since centrosome abnormalities are found in diverse malignancies and are associated with aberrant proliferation that is characteristic of carcinogenesis (39). CDK2 antagonism prevents centriole overduplication and reduces the extent of genomic instability while not disrupting centriole duplication and cell cycle progression (40–42). This study found that CDK2 antagonism also inhibits centrosome clustering via CP110. This is a way to target centrosomes. Pathways involved in centrosome clustering are usually dispensable in normal diploid cells since such cells, with a few exceptions like hepatocytes, do not have supernumerary centrosomes (39). This difference can be exploited therapeutically in aneuploid cancer cells versus normal diploid cells. Future translational research should explore this possibility in the context of clinical trials.
This study determined that KRAS mutations accelerate CP110 degradation rates by upregulating ubiquitin ligase SCFcyclinF. This sensitized lung cancer cells to CDK2 antagonism. This finding leads to the proposal that targeting lung cancers by upregulating CP110 degradation and inhibiting CDK2 activity would augment anti-neoplastic effects. Indeed, knockdown of the CP110 deubiquitinase USP33 along with CDK2 inhibition markedly increased anaphase catastrophe in human lung cancer cells, as in Fig. 6. This would overcome the potential limitation that lung cancers with reduced CP110 expression are particularly sensitive to CDK2 antagonism. Further studies are needed to determine the optimal ways to exploit anaphase catastrophe and centrosome functions for lung cancer therapy or prevention.
Seliciclib is an orally bioavailable inhibitor of CDK activity that reversibly competes for binding to the ATP pocket of the kinase catalytic subunit (19,43). Seliciclib prominently inhibits CDK-2, but affects CDK-1, CDK-7, and CDK-9 much less (19,43). Antitumor activity is reported against many human cancer cell lines, including those of breast, prostate, and lung cancer origins (19). Seliciclib has been tested in Phase I and Phase II clinical trials sponsored by Cyclacel Pharmaceuticals for treatments of non–small cell lung cancer (NSCLC) and nasopharyngeal carcinoma (43). Seliciclib is undergoing clinical testing in combination with the nucleoside analogue Sapacitabine (CYC682) (43) and with an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) (43).
Recently, roles of deubiquitinases in cancer biology and therapeutics were examined in multiple cancers and inhibitors designed to target deubiquitinases are being studied in oncology (44,45). Antitumor effects of inhibitors targeting USP7, USP8 and USP14 are being tested in different cancer models and preclinical results are promising (44,45).
In this study, USP33 knockdown was achieved as a proof-of-concept because USP33 can antagonize cyclin F actions on CP110 in regulating centrosome duplication, maturation and separation (24). USP33 is reported as down-regulated in human lung cancers and reduced USP33 expression is associated with an unfavorable clinical outcome (46). Yet, USP33 is upregulated in pancreatic cancers where centrosome amplification and KRAS mutations are often detected (24). Hence, more studies are needed to reveal cancer-type-specific roles of USP33 in human cancers.
Another promising strategy to sensitize lung cancers to CDK2 inhibition is by targeting the cyclin F degradation pathway. Cyclin F is necessary for maintaining chromosome stability and genome stability (22,47). Thus, up-regulating cyclin F should augment anti-tumor effects in combination therapies. Degradation of cyclin F is signaled through ATR by a mechanism that is not yet well understood (48). Further investigation of the degradation pathway of cyclin F is needed to identify potential targets that would increase cyclin F levels in cancer cells.
In summary, this study identified CP110 sites that mediate anaphase catastrophe and centrosome clustering following CDK2 inhibition. KRAS mutation can down-regulate CP110 protein expression by accelerating its degradation rate by upregulating the ubiquitin ligase SCFcyclinF. This study provided evidence for SCFcyclinF and USP33 affecting CP110 expression in human lung cancer cells. This in turn sensitized lung cancer cells towards CDK2 antagonism in a mechanism that is independent of CP110 phosphorylation. Thus, this is a tractable combination therapeutic approach. Combining CDK2 inhibition with repression of CP110 protein augments anti-neoplastic effects against human lung cancers. Taken together, the findings presented here exploit insights of the mechanism through which KRAS mutation can regulate CP110 expression. This helps elucidate the link between CP110, centrosome clustering and anaphase catastrophe. Future work should explore the relevance of these findings in the lung cancer clinic.
Supplementary Material
Acknowledgments
We thank members of the Dmitrovsky and Compton laboratories for their helpful consultation.
Financial Support: This work was supported by National Institutes of Health (NIH) and National Cancer Institute (NCI) grants R01-CA087546 (E. Dmitrovsky and S.J. Freemantle), R01-CA190722 (E. Dmitrovsky and S.J. Freemantle), and R37-GM051542 (D. Compton), by a Samuel Waxman Cancer Research Foundation (SWCRF) grant (E. Dmitrovsky and D. Compton), Mary Jo’s Fund to Fight Cancer (S.J. Freemantle), and by American Cancer Society Postdoctoral Fellowship PF-12-031-01-CCG (K. Godek). E. Dmitrovsky is an American Cancer Society Professor supported by a generous gift from the F.M. Kirby Foundation. Dartmouth’s Norris Cotton Cancer Center shared resources were used and were supported in part by NCI Grant SP30CA623108.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
References
- 1.Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122:1138–43. doi: 10.1172/JCI59954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowell PC. Clonal evolution of tumor-cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 4.Weaver BA, Cleaveland DW. The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell. 2008;14:431–433. doi: 10.1016/j.ccr.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci (USA) 2009;106:109–120. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 8.Liu D, Vader G, Vromans MJ, Lampson MA, Lens SM. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science. 2009;323:1350–1353. doi: 10.1126/science.1167000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi W, Liu X, Cooke LS, Persky DO, Miller TP, Squires M, Mahadevan D. AT9283, a novel aurora kinase inhibitor, suppresses tumor growth in aggressive B-cell lymphomas. Int J Cancer. 2012;130:2997–3005. doi: 10.1002/ijc.26324. [DOI] [PubMed] [Google Scholar]
- 10.Boss DS, Witteveen PO, van der Sar J, Lolkema MP, Voest EE, Stockman PK, et al. Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann Oncol. 2011;22:431–7. doi: 10.1093/annonc/mdq344. [DOI] [PubMed] [Google Scholar]
- 11.Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70:9846–54. doi: 10.1158/0008-5472.CAN-10-3001. [DOI] [PubMed] [Google Scholar]
- 12.Tsuboi K, Yokozawa T, Sakura T, Watanabe T, Fujisawa S, Yamauchi T, et al. A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk Res. 2011;35:1384–9. doi: 10.1016/j.leukres.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22(16):2189–203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watts CA, Richards FM, Bender A, Bond PJ, Korb O, Kern O, et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem Biol. 2013;20:1399–410. doi: 10.1016/j.chembiol.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67:2982–9. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 17.Mateos MV, Gutiérrez NC, Martín-Ramos ML, Paiva B, Montalbán MA, Oriol A, et al. Outcome according to cytogenetic abnormalities and DNA ploidy in myeloma patients receiving short induction with weekly bortezomib followed by maintenance. Blood. 2011;118:4547–53. doi: 10.1182/blood-2011-04-345801. [DOI] [PubMed] [Google Scholar]
- 18.Usmani SZ, Bona R, Li Z. 17 AAG for HSP90 inhibition in cancer–from bench to bedside. Curr Mol Med. 2009;9:654–64. doi: 10.2174/156652409788488757. [DOI] [PubMed] [Google Scholar]
- 19.Galimberti F, Thompson SL, Liu X, Li H, Memoli V, Green SR, et al. Targeting the cyclin E-Cdk-2 complex represses lung cancer growth by triggering anaphase catastrophe. Clin Cancer Res. 2010;16:109–20. doi: 10.1158/1078-0432.CCR-09-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu S, Danilov AV, Godek K, Orr B, Tafe LJ, Rodriguez-Canales J, et al. CDK2 inhibition causes anaphase catastrophe in lung cancer through the centrosomal protein CP110. Cancer Res. 2015;75:2029–38. doi: 10.1158/0008-5472.CAN-14-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell. 2002;3:339–50. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- 22.D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCFCyclin F controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–42. doi: 10.1038/nature09140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao J, Shen Y, Zhu L, Xu Y, Zhou Y, Wu Z, et al. miR-129-3p controls cilia assembly by regulating CP110 and actin dynamics. Nat Cell Biol. 2012;14:697–706. doi: 10.1038/ncb2512. [DOI] [PubMed] [Google Scholar]
- 24.Li J, D’Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature. 2013;495:255–9. doi: 10.1038/nature11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walentek P, Song R, He L. microRNAs and cilia: An ancient connection. Cell Cycle. 2014;13:2315–6. doi: 10.4161/cc.29827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song R, Walentek P, Sponer N, Klimke A, Lee JS, Dixon G, et al. miR-34/449 miRNAs are required for motile ciliogenesis by repressing CP110. Nature. 2014;510:115–20. doi: 10.1038/nature13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bärenz F, Hoffmann I. Cell Biology: Cell biology: DUBing CP110 controls centrosome numbers. Curr Biol. 2013;23:R459–60. doi: 10.1016/j.cub.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Kim S, Kobayashi T, Liang F-X, Korzeniewski N, Duensing S, et al. Neurl4, a novel daughter centriole protein, prevents formation of ectopic microtubule organizing centres. EMBO Rep. 2012;13:547–53. doi: 10.1038/embor.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, et al. Control of centriole length by CPAP and CP110. Curr Biol. 2009;19:1005–11. doi: 10.1016/j.cub.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 30.Tsang WY, Spektor A, Luciano DJ, Indjeian VB, Chen Z, Salisbury JL, et al. CP110 cooperates with two calcium-binding proteins to regulate cytokinesis and genome stability. Mol Biol Cell. 2006;17:3423–34. doi: 10.1091/mbc.E06-04-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsang WY, Bossard C, Khanna H, Peränen J, Swaroop A, Malhotra V, et al. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev Cell. 2008;15:187–97. doi: 10.1016/j.devcel.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell. 2007;130:678–90. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 33.Tsang WY, Spektor A, Vijayakumar S, Bista BR, Li J, Sanchez I, et al. Cep76, a centrosomal protein that specifically restrains centriole reduplication. Dev Cell. 2009;16:649–60. doi: 10.1016/j.devcel.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Tourneau C, Faivre S, Laurence V, Delbaldo C, Vera K, Girre V, et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur J Cancer. 2010;46:3243–50. doi: 10.1016/j.ejca.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Dragnev KH, Ma T, Cyrus J, Galimberti F, Memoli V, Busch AM, et al. Bexarotene plus erlotinib suppress lung carcinogenesis independent of KRAS mutations in two clinical trials and transgenic models. Cancer Prev Res. 2011;4:818–28. doi: 10.1158/1940-6207.CAPR-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66:4198–207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 37.Petty WJ, Li N, Biddle A, Bounds R, Nitkin C, Ma Y, et al. A novel retinoic acid receptor beta isoform and retinoid resistance in lung carcinogenesis. J Natl Cancer Inst. 2005;97:1645–51. doi: 10.1093/jnci/dji371. [DOI] [PubMed] [Google Scholar]
- 38.Pitha-Rowe I, Hassel BA, Dmitrovsky E. Involvement of UBE1L in ISG15 conjugation during retinoid-induced differentiation of acute promyelocytic leukemia. J Biol Chem. 2004;279:18178–87. doi: 10.1074/jbc.M309259200. [DOI] [PubMed] [Google Scholar]
- 39.Korzeniewski N, Hohenfellner M, Duensing S. The centrosome as potential target for cancer therapy and prevention. Expert Opin Ther Targets. 2013;17:43–52. doi: 10.1517/14728222.2013.731396. [DOI] [PubMed] [Google Scholar]
- 40.Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci USA. 2000;97:10002–7. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duensing S, Duensing A, Lee DC, Edwards KM, Piboonniyom SO, Manuel E, et al. Cyclin-dependent kinase inhibitor indirubin-3′-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene. 2004;23:8206–15. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- 42.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition Cancer Cell. 2003;3:233–45. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 43.Khalil HS, Mitev V, Vlaykova T, Cavicchi L, Zhelev N. Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J Biotechnol. 2015;202:40–9. doi: 10.1016/j.jbiotec.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 44.Edelmann MJ, Nicholson B, Kessler BM. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev Mol Med. 2011;13:e35. doi: 10.1017/S1462399411002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–58. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen P, Kong R, Liu J, Zhu L, Chen X, Li X, et al. USP33, a new player in lung cancer, mediates Slit-Robo signaling. Protein Cell. 2014;5:704–13. doi: 10.1007/s13238-014-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Angiolella V, Esencay M, Pagano M. A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013;23:135–40. doi: 10.1016/j.tcb.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–34. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.