

Abstract
HER2/neu gene amplification and PIK3CA driver mutations are common in uterine serous carcinoma (USC), and may represent ideal therapeutic targets against this aggressive variant of endometrial cancer. We examined the sensitivity to neratinib, taselisib and the combination of the two compounds in in vitro and in vivo experiments using PIK3CA mutated and PIK3CA-wild type HER2/neu amplified USC cell lines. Cell viability and cell cycle distribution were assessed using flow-cytometry assays. Downstream signaling was assessed by immunoblotting. Preclinical efficacy of single versus dual inhibition was evaluated in vivo using two USC-xenografts. We found both single agent neratinib and taselisib to be active but only transiently effective in controlling the in vivo growth of USC xenografts harboring HER2/neu gene amplification with or without oncogenic PIK3CA mutations. In contrast, the combination of the two inhibitors caused a stronger and long lasting growth inhibition in both USC xenografts when compared to single agent therapy. Combined targeting of HER2 and PIK3CA was associated with a significant and dose-dependent increase in the percentage of cells in the G0/G1 phase of the cell cycle and a dose-dependent decline in the phosphorylation of S6. Importantly, dual inhibition therapy initiated after tumor progression in single agent-treated mice was still remarkably effective at inducing tumor regression in both large PIK3CA or pan-ErbB inhibitor-resistant USC xenografts. Dual HER2/PIK3CA blockade may represent a novel therapeutic option for USC patients harboring tumors with HER2/neu gene amplification and mutated or wild type PIK3CA resistant to chemotherapy.
Keywords: Uterine serous carcinoma, HER2, PIK3CA, neratinib, taselisib
Introduction
Endometrial cancer is the most common gynecologic malignancy with approximately 54,870 new cases and 10,170 estimated deaths related to the disease in the United States annually (1). Recently, using a comprehensive genetic investigation, The Cancer Genome Atlas (TCGA) Research Network provided compelling evidence that endometrial cancers result from heterogeneous somatic mutations and classified endometrial cancers into four categories: 1) polymerase epsilon (POLE)-ultramutated, 2) microsatellite instability hypermutated, 3) copy-number low and 4) copy-number high, serous-like (2). In this landmark study, patients harboring uterine serous carcinoma (USC) were found to have the worst prognosis as compared with all the other groups of endometrial cancer (2). USC is high grade by definition. Because of its biologic aggressiveness, early stage USC (i.e., Stage I) is treated after surgical staging with systemic cytotoxic chemotherapy with or without localized radiation therapy (3–5). Unfortunately, about 70% of patients have extra-uterine metastases at the time of initial staging and up to 50% of patients treated with surgery will develop recurrent disease, which is fatal in the majority of the cases (5, 6). The development of novel and more effective treatment modalities remain an unmet medical need in USC patients.
Human epidermal growth factor receptor 2 (HER2) is a member of the human epidermal growth factor receptor superfamily that consists of three additional tyrosine kinase receptors (HER1/EGFR, HER3 and HER4) (7). Unlike the other epidermal growth factor receptors, HER2 has no known ligand and functions as a preferred partner for heterodimerization with any of the other members of the EGF receptor family and thus plays an important role in the coordination of the complex HER2/neu signaling network that is responsible for regulating cell growth and differentiation (7). Amplification of the HER2 (ERBB2) gene has been described in many human malignancies including but not limited to breast, colon and gastric cancer and it has been reported in up to 35% of USC (8–11). Moreover, early reports have demonstrated that HER2 protein overexpression and gene amplification is associated with more aggressive disease, worse prognosis and resistance to therapy in multiple human tumors including USC (11–13).
The phosphatidylinositol-3-kinase (PI3KCA) gene encodes for a heterodimeric protein with an 85-kDa regulatory subunit (PI3KR1) and a 110-kDa catalytic subunit (PI3KCA). PI3K pathway is known to play a fundamental role in cellular functions including proliferation, survival and growth in normal as well as neoplastic cells. Importantly, the catalytic subunit of the PIK3CA gene is frequently mutated or amplified in the different types of endometrial cancers and may therefore represent an attractive target for the development of novel, potentially effective therapies against biologically aggressive tumors such as USC (14–21).
Neratinib, (HKI-272, Puma Biotechnology, Los Angeles) is an oral, potent and irreversible inhibitor of EGFR, HER2 and HER4 tyrosine kinases with promising preclinical activity against HER2-overexpressing cell lines (22). Importantly, neratinib has been demonstrated to be significantly more effective when compared to the first generation (i.e., reversible) EGFR and HER2 inhibitors (22–25), and it is currently in Phase III trials in breast cancer patients (NCT01808573). Taselisib, (GDC-0032, Genentech, South San Francisco, CA), is a novel, oral, selective inhibitor of PIK3CA. Taselisib binds the ATP-binding pocket of PI3K with selective preference for the mutated form of PIK3CA (26) and it is currently tested in Phase II/III clinical trials against multiple human tumors (i.e., NCT02154490).
In this study, we have evaluated the effect of single vs dual HER2/PIK3 inhibition in multiple FISH±/PIK3CA wild type and FISH±/PIK3CA mutated primary USC cell lines fully characterized by whole exome sequencing (20). We demonstrate for the first time that the dual-targeting of HER2 and PIK3CA with neratinib and taselisib is highly synergistic against HER2/neu amplified PIK3CA mutated and PIK3CA wild type USC primary cell lines in vitro as well as in vivo and able to overcome single agent resistance in USC xenografts progressing on single agent therapy.
Materials and Methods
USC cell lines and inhibitors
Study approval was obtained from the Institutional Review Board at Yale University, and all patients signed consent prior to tissue collection according to the institutional guidelines. Four primary USC cell lines authenticated by whole exome sequencing (WES) were established from chemotherapy-naïve patients at the time of primary staging surgery after sterile processing of fresh tumor biopsy samples, as described previously and evaluated in our study (20). Source-patient characteristics of the USC cell lines are described in Table 1. HER2/neu gene amplification in the cell lines was evaluated by fluorescence in situ hybridization (FISH) and has been previously been reported (20). Neratinib and taselisib (purchased from LC Laboratories Woburn, MA and Medchemexpress, NJ, respectively) were dissolved in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) as a 10 mM stock solution and diluted in culture medium immediately before use. USC primary cell lines with limited in vitro passages (i.e., #10) were used in the experiments described below.
Table 1.
USC cell lines characteristics
| CELL LINES ID |
AGE (YEARS) |
RACE | FIGO STAGE |
YEAR OF DIAGNOSIS |
PIK3CA MUTATIONS |
HER2/neu FISH |
HER2/neu IHC |
NERATINIB IC50 (µM) |
TASELISIB IC50 (µM) |
|---|---|---|---|---|---|---|---|---|---|
| USPC-ARK-1 | 62 | AA | IVA | 1997 | E542K | AMPLIFIED | 3+ | 0.023 | 0.014 |
| USPC-ARK-2 | 63 | AA | IVB | 1998 | NOT DETECTED | AMPLIFIED | 3+ | 0.013 | 0.049 |
| USPC-ARK-20 | 42 | C | II | 1999 | H1047R | AMPLIFIED | 3+ | 0.012 | 0.041 |
| USPC-ARK-21 | 70 | C | IA | 2012 | NOT DETECTED | AMPLIFIED | 3+ | 0.010 | 0.070 |
FIGO, International Federation of Gynecology and Obstetrics stage; AA= African-American; C= Caucasian.
Cell viability assay and synergism
The effect of single agent neratinib and taselisib on the viability of 3 primary USC cell lines (i.e., USPC-ARK-2, 20, 21) has been previously reported (23, 27). The effect of neratinib on the viability and IC50 of USPC-ARK-1 cell line was determined in flow-cytometry based assays as previously described (23, 27). Briefly, tumor cells were plated in six-well tissue culture plates and treated with neratinib at concentrations of 0.005, 0.01, 0.05, 0.1 and 0.5 µM 24 hrs after plating. After 72 hours of additional incubation, well contents were harvested in their entirety, centrifuged and then stained with propidium iodide (2 µL of a 500 µg/mL stock solution in PBS) for flow cytometric counts. The viable cells were then quantified using flow-cytometry (FACSCalibur, Beckton-Dickinson, San Jose, CA) as percentage of viable cells (mean ± standard error (SEM)) after exposure to different concentrations of neratinib relative to vehicle-treated cells (i.e., 100% viable). After having determined the IC50 dose of neratinib for the USPC-ARK-1, all four cell lines were treated with neratinib and taselisib (0.005 µM, 0.01 µM and 0.02 µM) as single agent or in 1:1 combination. After 72 hrs of incubation, cell proliferation was assessed as previously described (23, 27). A minimum of 3 independent experiments per USC cell line was performed. For drug combination studies, the synergistic effect was assessed by the combination index (CI), according to the method of Chou & Talalay where synergism is defined as CI < 1, while antagonism is CI > 1, and an additive effect is considered as CI = 1 (at affected fraction (Fa)= 0.50, 0.75, 0.90 and 0.95). The CI values were calculated using CompuSyn (ComboSyn, Inc.) as previously described (28).
Flow-cytometry analysis of cell cycle in primary USC cell lines
After 24 hrs exposure to neratinib (0.01 µM), taselisib (0.01 µM) and the combination of both drugs, treated and control cells were permeabilized with ice-cold 70% ethanol and fixed for 30 min at 4° C. After spinning at 2000 rpm for 5 min and discarding supernatant, cells were suspended in 1 ml of PBS. After additional spinning at 2000 rpm for 5 min, 100 µl of ribonuclease (100 µg/ml, DNase free, Sigma) was added for 5 min incubation at room temperature, before exposure to 400 µl of propidium iodide (50 µg/ml in PBS). Treated and untreated control cells were acquired with FACSCalibur, using Cell Quest software (BD Biosciences, San Jose, CA) and were analyzed using Flowjo software (Ashland, OR).
Immunoblotting
Cells were seeded in Petri tissue culture plates (100,000 cells) and left to adhere overnight. Cells were then treated with 0.01 µM of neratinib, taselisib and the combination of both for 24–48 hours. After incubation, cells were washed once with PBS, mechanically scraped and lysed for 30 minutes on ice with 75 µL of radioimmunoprecipitation assay (RIPA) buffer (50 mmol/L Tris-HCl, pH 8, 150 mmol/L NaCl, Triton X-100 1%, Na deoxicolate 0.5%, SDS 0.1%, MgCl 5mmol/L in H2O) supplemented with protease inhibitor cocktail (#78441, Thermo Scientific, Rockford, IL). Lysates were prepared by collecting supernatants after centrifugation for 10 minutes at 10,000 × g at 4° C. Protein levels were quantified with BCA Protein Assay Kit (#23225, Thermo Scientific, Rockford, IL), and equal amounts were loaded for SDS-PAGE using 4% to 20% acrylamide precast gels (Bio-Rad), followed by transfer to polyvinylidene difluoride membranes (Bio-Rad). Antibodies used for immune detection of proteins were pHER2 (#2243, Cell Signaling Technology, Inc.), pEGFR (#2234, Cell Signaling Technology, Inc.), pS6 (#4856, Cell Signaling Technology, Inc.), pAKT (#4060, Cell Signaling Technology, Inc.), pERK (#4376, Cell Signaling Technology, Inc.) and GAPDH (#2118, Cell Signaling Technology, Inc). Incubation of the membranes with primary antibodies was carried overnight in 3% BSA in PBS-Tween at 4°C. After incubation, membranes were washed three times with 1% milk in PBS-Tween (0.2%) at room temperature and incubated with an HRP-linked secondary antibody (#7074, Cell Signaling Technology, Inc) in 5% milk PBS-Tween (0.2%) for one hour before washing four times in 1% milk PBS-Tween. Signals were detected with Western blotting detection reagents (Thermo Scientific, Rockford, IL). Bands were then visualized and the blots developed using an enhanced chemiluminescent system (GEL Logic 1500, Carestream Health, Rochester, NY)
Flow-cytometry analysis of phosphorylated S6 intracellular levels
Next we evaluated pS6 expression levels by flow-cytometry. Cells were plated in a 6-well plate (40.000 cells per well). After 24 hrs exposure to 0.01 µM of taselisib, 0.01 µM of neratinib, and the combination of both agents, treated and untreated control cells were fixed in 4% formaldehyde and permeabilized with ice-cold 90% methanol. Treated and untreated control cells were incubated with primary rabbit monoclonal antibody against pS6 (# 4856, Cell Signaling Technology, Inc., Danvers, MA) following the protocol provided by the manufacturers and stained with a fluorescein isothiocyanate-conjugated goat anti-rabbit F(ab’)2 immunoglobulin as a secondary reagent (Chemicon International, Temecula, CA). Cells (i.e., 5,000 events per sample) were analyzed on FACSCalibur, using Cell Quest software.
In vivo assay of drug effect
A representative FISH± cell line (USPC-ARK-2) and a representative FISH±/PIK3CA-mutated cell line (USPC-ARK-1) were injected into the subcutaneous region of 5–8 week old SCID mice (Harlan Laboratories, Indianapolis, IN). After implantation of cells, tumors were monitored until they reached a tumor volume of 0.1 cm3 prior to initiating the treatment. Mice were then randomized into the following 4 groups: vehicle (0.5% methylcellulose-0.2% Tween 80), taselisib (10 mg/kg), neratinib (40mg/kg) and the combination of taselisib (10 mg/kg) plus neratinib (40mg/kg). Each group consisted of 5 mice. Treatments were given orally once a day, 5 days a week for 60 days after which mice were euthanized according to the rules and regulations set forth by the Institutional Animal Care and Use Committee (IACUC). Tumor volume was calculated by the formula V= length × (width)2 × 0.5. Tumor sizes and body weights were recorded twice per week. Tumors volumes are plotted as means ± SEM.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism5 version 6 (GraphPad Software, Inc., San Diego, CA). For each independent experiment of neratinib on USPC-ARK-1, the measures of growth under different dose levels were normalized to the mean of the control group receiving no drug, so that all data were expressed as a proportion of the control. Normalized data were then fit via nonlinear regression to a normalized logistic response curve against the base-10 logarithms of dose in M, and the resulting parameter estimates were used to calculate the value of the IC50 (in log10 units) for that experiment. One-way ANOVA was used to determine the statistical significance of the effects of combination treatment on the different cell lines in vitro when compared to the control and to the single agent treatment. For the flow-cytometry experiments, changes in the phosphorylated S6 protein levels were analyzed comparing the mean intensity of fluorescence (MFI) before and after the exposure to the inhibitors. The measures of the MFIs under different dose levels were normalized to the mean of the control group receiving no drug, so that all data were expressed as a proportion of the control. Unpaired t-test was used to assess pS6 changes and cell cycle changes in the HER2/neu amplified PIK3CA mutated or wild type cell lines. Statistical differences between mean tumor volumes at specific time points were performed using an unpaired t-test. Error bars represent SEM. Differences in all comparisons were considered significant at P values < 0.05.
Results
In vitro activity of neratinib and taselisib in HER2/neu gene amplified cell lines
First, we evaluated the sensitivity of the USPC-ARK-1 cell line to neratinib. We found strong growth inhibition using increasing concentrations of neratinib with a mean inhibitory concentration (IC50) ± (SEM) of 0.023 ± 0.008 µM (Figure S1). We next evaluated the effect of neratinib, taselisib and the combination of both in all four cell lines at the IC50 concentrations identified in previous studies (23, 27). As shown in Figure 1, the combination of the two inhibitors was able to induce a more potent and highly significant cell growth inhibition when compared to single agent therapy in all the cell line tested. To further confirm these results we evaluated the synergism between the two compounds in all cell lines tested following combination treatment at multiple paired concentrations. Data were evaluated for potential synergistic activity (CI values) using the software CompuSyn. In all four HER2/neu gene amplified cell lines tested the combination of neratinib and taselisib showed synergistic activity. Results are shown in Table 2. For the USPC-ARK-1 cell line the following CI values were obtained: 0.48753 (Fa=0.50), 0.24043 (Fa=0.75), 0.11857 (Fa=0.90) and 0.07332 (Fa=0.95). For the USPC-ARK-2 cell line the CI values were: 0.46474 (Fa=0.50), 0.32323 (Fa=0.75), 0.25874 (Fa=0.90) and 0.23128 (Fa=0.95). For the USPC-ARK-20 cell line the CI values were: 0.72491 (Fa=0.50), 0.60347 (Fa=0.75), 0.64375 (Fa=0.90) and 0.67345 (Fa=0.95). For the USPC-ARK-21 cell line the CI values were: 0.70324 (Fa=0.50), 0.65658 (Fa=0.75), 0.59501 (Fa=0.90) and 0.55665 (Fa=0.95).
Figure 1.

Cell viability assay of the four USC cell lines treated with neratinib, taselisib and the combination of both at the indicated concentration for 72 hrs. Cell viability was analyzed by flow cytometry and was normalized to the mean of the control group receiving no drug, so that all data were expressed as a proportion of the control. Data are expressed as mean ± SEM from three independent experiments (*=P<0.05 when compared to the control, to neratinib and to taselisib)
Table 2.
Combination index of the combination of neratinib and taselisib in USC cell lines
| CELL LINES ID | CI | DESCRIPTION |
|---|---|---|
| USPC-ARK-1 | 0.11857 | Synergism |
| USPC-ARK-2 | 0.25874 | Synergism |
| USPC-ARK-20 | 0.64375 | Synergism |
| USPC-ARK-21 | 0.59501 | Synergism |
Note: USC cell lines were treated with increasing concentration of neratinib, taselisib or the combination. Viability was evaluated after 72 hrs of incubation by flow cytometry and CI was calculated using CompuSyn (Fa= 0.9). CI <1 indicates synergy, CI < 0.3 indicates strong synergy, CI < 0.1 indicates very strong synergy (Ref 28).
Cell cycle analysis
Next, we investigate cell cycle progression after treatment with neratinib, taselisib and the combination of both inhibitors in two representative FISH±/PIK3CA mutated or wild type cell lines (USPC-ARK-2 and USPC-ARK-1). In USPC-ARK-2 (i.e., PIK3CA wild type) taselisib as single agent, was unable to significantly delay cell cycle (increasing the G1 phase) after 24 hrs exposure. In contrast, neratinib was effective in inducing a significant increase in the percentage of the cells blocked in the G0/G1 phase (P=0.01, Figure 2). However, when we analyzed the effect of the combination of the two inhibitors, we found a significant increase in the percentage of cells blocked in the G1 phase of the cell cycle when compared to untreated controls or cells treated with the single agent neratinib and taselisib (P=0.002, P=0.03 and P=0.003 respectively, Figure 2). Next we evaluated cell cycle analysis in USPC-ARK-1 (i.e., PIK3CA mutated). We found both inhibitors, (i.e., neratinib and taselisib), to be able to increase the number of cells blocked in G0/G1 phase when compared to control cells (P=0.007 and P=0.03 respectively, Figure 2). Furthermore, we found the combination to induce a significant increase in the percentage of the cells blocked in the G1/G0 phase of the cell cycle when compared to the untreated control, to neratinib and taselisib used as single agents (P=0.0006, P=0.01 and P=0.001 respectively, Figure 2)
Figure 2.

Graph representing mean ± SEM of percentages of cells in G0/G1, after exposure to neratinib, taselisib and the combination of both at the indicated concentration in a representative FISH±\PIK3CA mutated USC cell line (USPC-ARK-1) and in a representative FISH±\PIK3CA wild type USC cell line (USPC-ARK-2).
Immunoblotting
Western blotting analyses of phosphorylated HER2, EGFR, AKT and S6 were performed after 24–48 hours of treatment at the selected drug concentrations described in Methods section in USPC-ARK-2 and USPC-ARK-1. We found neratinib to be able to reduce the levels of p-HER2 and p-EGFR in both cell lines tested (Figure 3A). Taselisib single agent was active in reducing pAKT in USPC-ARK-1 but not USPC-ARK-2 cell lines while combination treatment led to reduced levels of p-AKT and p-S6 in USPC-ARK-1 cells, and a decrease in p-S6 but not p-AKT in USPC-ARK-2 treated cells (Figure 3A). Data presented in Figure 3B (i.e., 48 hrs after exposure to drugs) and S2 further confirm that the combination of the two inhibitors was able to induce a significant dephosphorylation of S6 when compared to control cells in both cell lines tested. In additional experiments we performed western blot analyses of phosphorylated HER2, EGFR, AKT and ERK in the USPC-ARK-1 primary cell line after two weeks exposure to single agent neratinib or Taselisib (i.e., single agent resistant cell lines). These cells were confirmed to be resistant in vitro in proliferation assays to both single agent taselisib and neratinib but highly responsive to the drug combination (Supplementary Figure S3). We found a significant increase in phosphorylated AKT and a consistent reduction in the levels of p-HER2 and p-EGFR after prolonged exposure to neratinib (Figure 3 C). In contrast, we found a significant increase in phosphorylated HER2, EGFR and ERK after two weeks exposure to taselisib.
Figure 3.
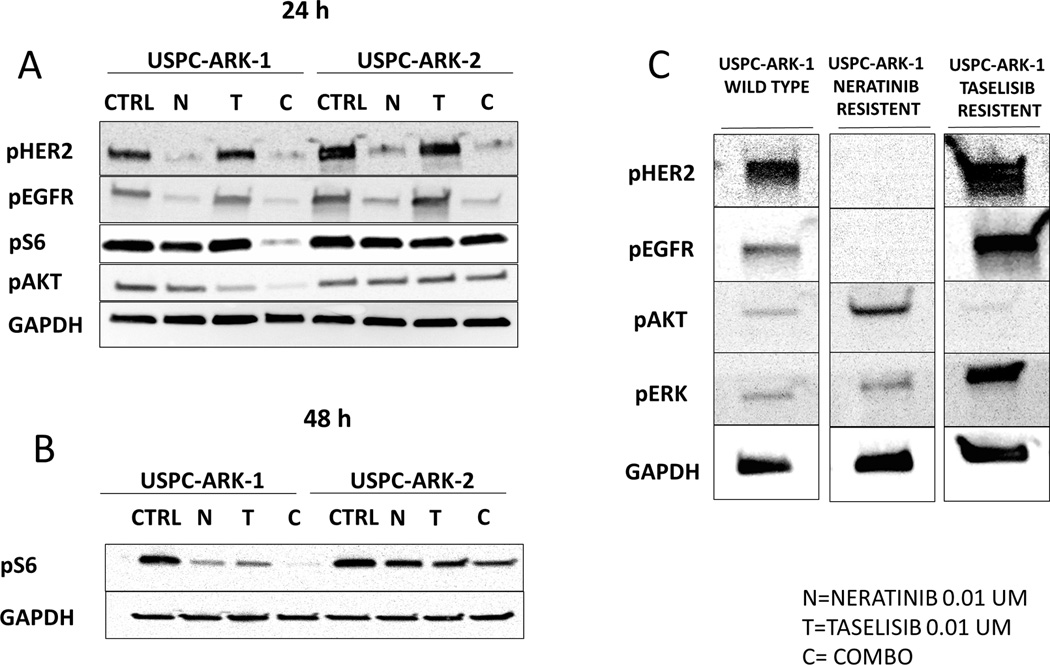
A: Two representative cell lines were treated with neratinib 0.01 µM, taselisib 0.01 µM and the combination of both (neratinib 0.01 µM and taselisib 0.01 µM) collected 24 hours and 48 h (B) after treatment. Cells were used for Western blotting analyses of phosphorylated HER2, EGFR, AKT and S6. Neratinib was able to reduce levels of p-HER2 and p-EGFR in both cell lines tested. Combination treatment led to reduced levels of p-AKT and p-S6 in USPC-ARK-1 cells, while a decrease in p-S6 but not p-AKT was found in combination-treated USPC-ARK-2 cells. Expression of GAPDH was used as loading control. C: USPC-ARK-1 single agent resistant cell lines. Two weeks exposure to neratinib induced significant increase in phosphorylated AKT, while two weeks exposure to taselisib induced a significant increase phosphorylated HER2, EGFR and ERK.
Antitumor activity of neratinib and taselisib in xenograft models
To validate our in vitro results, we next determined the in vivo activity of neratinib, taselisib and the combination of the two drugs in two different animal models. First we evaluated the tumor growth of USPC-ARK-1 (FISH±/ PIK3CA mutated) xenograft under the therapeutic conditions described above. Consistent with the in vitro data, both single agent neratinib and taselisib were able to induce a significant tumor growth inhibition (after 4 and 14 days of treatment, respectively) when compared to the vehicle group (P=0.01 and P=0.03 respectively, Figure 4, upper panel). Furthermore, the combination of the compounds was able to induce tumor regression after 4 days of treatment when compared to the control (P<0.0001) and to taselisib (P=0.01). Starting at 11 days after the beginning of treatment, the combination of the two inhibitors was able to induce a remarkable tumor growth inhibition when compared to single agent neratinib (P=0.01). This effect lasted for the entire treatment period (i.e., 60 days) after which the animals were euthanized. To determine whether the in vivo acquired resistance to single agent therapy was potentially reversible in the animal model, we also started a combination treatment adding the missing inhibitor to the neratinib or the taselisib group when the mean tumor volume reached 0.9 cm3. As shown in Figure 4 the combination was highly effective in inducing tumor regression in both groups of animals on single agent therapy. Next, we evaluated the in vivo activity of neratinib, taselisib and the combination of the two drugs in the USPC-ARK-2 xenografts (FISH±/PIK3CA wild type) under the same therapeutic conditions. Once again, both single agent neratinib and taselisib were able to induce a significant tumor growth inhibition (after 4 and 11 days of treatment, respectively) when compared to the vehicle group (P=0.005 and P=0.02 respectively, Figure 4). The combination of the compounds was able to induce tumor regression after 4 days of treatment when compared to the control (P=0.01). Importantly, similarly to USPC-ARK-1 cell line, the combination of the two inhibitors was also able to overcome single agent acquired resistance and induced remarkable tumor regression in both group of animals in progression during neratinib or taselisib single agent treatment (Figure 4, lower panel).
Figure 4.

In vivo treatment of USPC-ARK-1 and USPC-ARK-2. Mice were treated with vehicle control (0.5% methylcellulose-0.2% Tween 80), neratinib (40 mg\kg), taselisib (10 mg\kg) or the combination for 60 days. Measurements are reported as mean ± SEM. Upper panel: Graph showing a statistically significant difference in tumor growth of USPC-ARK-1 between the control group and the treated groups. Lower panel: Graph showing a statistically significant difference in tumor growth of USPC-ARK-2 between the control group and the treated groups. The arrows denote the time point in which we started the combination treatment in single agent resistant mice
Discussion
Patients diagnosed with advanced or recurrent biologically aggressive endometrial cancers such as USC have an extremely poor prognosis. The development of novel, effective therapies against USC resistant to chemotherapy remain desperately needed. In the last few years multiple comprehensive genetic studies from our group, the TCGA network as well as others have reported the mutational landscape of USC, giving the opportunity for identification of multiple deranged pathways as potential novel targets for the treatment of this highly lethal tumor (2, 20, 21, 29, 30). Because these comprehensive studies found that a large number of USC patients harbor alterations in the HER2/neu and/or the PIK3CA gene (20, 31, 32), the HER2/PI3K/AKT/mTOR pathway may represent a highly attractive therapeutic target against these rare tumors (31).
Consistent with this view, in the last few years several potent and highly selective compounds against the HER2/neu, PI3K, AKT or mTOR pathways have been generated. However, only limited amounts of preclinical data are currently available about the use of HER2-targeted agents or PI3K TKIs against USC (23, 27, 31). In addition, while most of the preclinical studies showed promising result using pan-ErbB or PI3K/mTOR inhibitors as single agents in mice engrafted with HER2 FISH± or HER2 FISH±/PIK3CA mutated cell lines, none of these studies was able to demonstrate durable tumor growth inhibition in vivo (23, 27, 33). Consistent with these preclinical results, emerging clinical data have so far shown limited single-agent activity of these inhibitors at tolerated doses in endometrial cancer (34–36).
The high genomic instability and heterogeneity of USC (2, 10, 20), combined with the improved understanding of the mechanism of USC carcinogenesis have recently provided a new alternative of using targeted agents against HER2, PI3K, AKT or mTOR in combination. Accordingly, in this study we have evaluated the efficacy of neratinib and taselisib as single agents and in combination against multiple genetically well characterized (i.e., whole exome sequenced) USC cell lines in vitro and in vivo. Similar to our previously reported findings (23, 27, 33), we found both single agent neratinib and taselisib to be highly active in vitro but only transiently effective in vivo in controlling the growth of both USC xenograft models harboring HER2/neu gene amplification with or without oncogenic PIK3CA mutations. These in vivo results in USC xenografts using an irreversible pan-ErbB or PI3K inhibitor are therefore consistent with the results of clinical studies in B-RAF mutated melanoma or EGFR mutated lung cancer patients treated with vemurafenib or gefitinib, respectively. In these studies, treatment with highly targeted agents resulted in an initial tumor shrinkage in the short-term (37, 38). Long term follow up suggested that patients who initially responded, tended to have progression typically only few months after starting treatment (39–41). Taken together these clinical results combined with our preclinical data in USC suggest that even in molecularly selected patients with well documented oncogenic “driver” gene mutations, the rapid acquisition of resistance may represent a significant barrier to the long term survival of cancer patients.
Preclinical studies in both breast and lung cancer cell lines and mouse models suggest that small molecule mediated inhibition of the HER2 alone is insufficient for complete inhibition of PI3K/mTOR activity, which may contribute to both primary and acquired resistance via persistent over-activation of downstream signaling (42–44). Consistent with this view, previous in vivo models suggest the addition of a mTOR inhibitor to a HER2 inhibitor may result in synergistic tumor growth inhibition and regression (45, 46). The combination of these two drug classes was similarly synergistic in vitro, with combinatorial suppression of the PI3K-mTOR-S6 axis. Furthermore, the addition of HER2 inhibition to PI3K/mTOR inhibition may potentially block feedback activation of PI3K–AKT/protein kinase B and extracellular signal regulated kinases that occur after mTOR inhibition alone (45, 46). Accordingly, when the efficacy of the combination of PI3K and HER2 inhibition was tested in HER2/neu amplified cell lines, we found a synergistic effect of the dual-inhibition in all USC cell lines tested (i.e., 4 out of 4) in vitro, with a more potent delay of the cell cycle and a more potent dephosphorylation of several downstream elements of the HER2/PI3K/mTOR pathway when compared to single agent therapy (Figures 3 and S2). More strikingly, the association of neratinib and taselisib in vivo in both USC xenograft model available was highly synergistic and effective in preventing tumor outgrowth for the entire duration of the in vivo study (i.e., 2 months). Moreover, the dual targeting of this pathway was also able to overcome the in vitro (Supplementary Figure S3) and in vivo acquired resistance to single agent neratinib or taselisib in both tumor models. These latter results showing high sensitivity to taselisib (i.e., PI3K inhibitor) or neratinib (i.e., pan-ErbB inhibitor) in single agent resistant tumors may potentially be explained by the results of our western blot experiments showing a significant increase in the phosphorylation of AKT (i.e., a downstream effector of PI3K) after prolonged exposure to neratinib as well as a significant increase in the phosphorylation of HER2, and EGFR (i.e., neratinib targets) after prolonged exposure to taselisib. Our molecular results in USC are therefore in agreement with the results of Serra et al., in breast cancer cell lines overexpressing HER2 who also found that combined administration of PI3K inhibitors with HER2 inhibitors resulted in superior anti-tumor activity when compared with single agent PI3K inhibitors as well as a compensatory activation of the downstream signaling pathways (47).
One possible limitation of dual therapy in the clinical setting is the potential increased toxicity as a result of combining two highly targeted agents. It is therefore worth noting that no evidence of increased in vivo acute or chronic toxicity was detected in our study. Indeed, no significant variation in behavior or body weights was found in mice receiving combination treatment when compared to the mice in the control groups (Supplementary figure S4). These data suggest that HER2 amplified USC resistant to neratinib or taselisib single agent therapy may be responsive to combinatorial treatment. These results could have important implications for patients currently being treated.
In conclusion, while treatment with targeted therapeutics may initially lead to dramatic tumor regression, cancers seem to invariably acquire resistance to these drugs. Our study represents the first preclinical demonstration that synergistic dual-targeting HER2/PIK3CA with neratinib and taselisib is able to achieve durable regression of established USC xenografts in vivo. Daily oral administration of the two compounds may represent a novel, potentially highly effective therapeutic strategy against HER2/neu amplified USC harboring mutated or wild type PIK3CA genes unresponsive to chemotherapy.
Supplementary Material
Acknowledgments
FINANCIAL SUPPORT: This work was supported in part by R01 CA154460-01 and U01 CA176067-01A1 grants from NIH, the Deborah Bunn Alley Foundation, the Tina Brozman Foundation, the Discovery to Cure Foundation and the Guido Berlucchi Foundation to A.D. Santin. This investigation was also supported by NIH Research Grant CA-16359 from the NCI and by The Italian Ministry of Health Grant RF-2010-2313497 to A.D. Santin.
Footnotes
CONFLICTS OF INTEREST: The authors report no conflicts of interest
References
- 1.Siegel RL, Miller KD, Jemal A. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrickson M, Ross J, Eifel P, Martinez A, Kempson R. Uterine papillary serous carcinoma: a highly malignant form of endometrial adenocarcinoma. Am J Surg Pathol. 1982;6:93–108. doi: 10.1097/00000478-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Moore KN, Fader AN. Uterine papillary serous carcinoma. Clin Obstet Gynecol. 2011;54:278–291. doi: 10.1097/GRF.0b013e318218c755. [DOI] [PubMed] [Google Scholar]
- 5.Fader AN, Boruta D, Olawaiye AB, Gehrig PA. Uterine papillary serous carcinoma: epidemiology, pathogenesis and management. Curr Opin Obstet Gynecol. 2010;22:21–29. doi: 10.1097/GCO.0b013e328334d8a3. [DOI] [PubMed] [Google Scholar]
- 6.Goff BA, Kato D, Schmidt RA, Ek M, Ferry JA, Muntz HG, et al. Uterine papillary serous carcinoma: patterns of metastatic spread. Gynecol Oncol. 1994;54:264–268. doi: 10.1006/gyno.1994.1208. [DOI] [PubMed] [Google Scholar]
- 7.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 8.Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353:1652–1654. doi: 10.1056/NEJMp058197. [DOI] [PubMed] [Google Scholar]
- 9.Geng Y, Chen X, Qiu J, Zhou Y, Wang J, Liu L, et al. Human epidermal growth factor receptor-2 expression in primary and metastatic gastric cancer. Int J Clin Oncol. 2014;19:303–311. doi: 10.1007/s10147-013-0542-9. [DOI] [PubMed] [Google Scholar]
- 10.Buza N, Roque DM, Santin AD. HER2/neu in Endometrial Cancer: A Promising Therapeutic Target With Diagnostic Challenges. Arch Pathol Lab Med. 2014;138:343–350. doi: 10.5858/arpa.2012-0416-RA. [DOI] [PubMed] [Google Scholar]
- 11.Buza N, English DP, Santin AD, Hui P. Toward standard HER2 testing of endometrial serous carcinoma: 4-year experience at a large academic center and recommendations for clinical practice. Mod Pathol. 2013;26:1605–1612. doi: 10.1038/modpathol.2013.113. [DOI] [PubMed] [Google Scholar]
- 12.Santin AD, Bellone S, Van Stedum S, Bushen W, Palmieri M, Siegel ER, et al. Amplification of c-erbB2 oncogene: a major prognostic indicator in uterine serous papillary carcinoma. Cancer. 2005;104:1391–1397. doi: 10.1002/cncr.21308. [DOI] [PubMed] [Google Scholar]
- 13.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 14.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 15.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 16.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 17.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009;106:4834–4839. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst. 2012;104:1503–1513. doi: 10.1093/jnci/djs345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 23.Schwab CL, English DP, Roque DM, Bellone S, Lopez S, Cocco E, et al. Neratinib shows efficacy in the treatment of HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol. 2014;135:142–148. doi: 10.1016/j.ygyno.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Wissner A, Mansour TS. The development of HKI-272 and related compounds for the treatment of cancer. Arch der Pharm. 2008;341:465–477. doi: 10.1002/ardp.200800009. [DOI] [PubMed] [Google Scholar]
- 25.Kosaka T, Yamaki E, Mogi A, Kuwano H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J Biomed Biotechnol. 2011;2011:165214. doi: 10.1155/2011/165214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597–4610. doi: 10.1021/jm4003632. [DOI] [PubMed] [Google Scholar]
- 27.Lopez S, Schwab CL, Cocco E, Bellone S, Bonazzoli E, English DP, et al. Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol. 2014;135:312–317. doi: 10.1016/j.ygyno.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 29.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 31.English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther. 2013;17:85–99. doi: 10.1007/s40291-013-0024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwab CL, Bellone S, English DP, Roque DM, Lopez S, Cocco E, et al. Afatinib demonstrates remarkable activity against HER2-amplified uterine serous endometrial cancer in vitro and in vivo. Br J Cancer. 2014;111:1750–1756. doi: 10.1038/bjc.2014.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan J, Yu Q. Molecular mechanisms of tumor resistance to PI3K-mTOR-targeted therapy. Chin J Cancer. 2013;32:376–379. doi: 10.5732/cjc.012.10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavlidou A, Vlahos NF. Molecular alterations of PI3K/Akt/mTOR pathway: a therapeutic target in endometrial cancer. Scientific World Journal. 2014;2014:709736. doi: 10.1155/2014/709736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin-Liberal J, Larkin J. Vemurafenib for the treatment of BRAF mutant metastatic melanoma. Future Oncol. 2015;11:579–589. doi: 10.2217/fon.14.252. [DOI] [PubMed] [Google Scholar]
- 38.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 39.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 40.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer. 2010;9:75. doi: 10.1186/1476-4598-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21:255–262. doi: 10.1093/annonc/mdp304. [DOI] [PubMed] [Google Scholar]
- 43.PIK3CA mutation predicts resistance to breast cancer therapy. Cancer discovery. 2014;4:OF3. doi: 10.1158/2159-8290.CD-NB2013-180. [DOI] [PubMed] [Google Scholar]
- 44.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 45.O'Brien NA, McDonald K, Tong L, von Euw E, Kalous O, Conklin D, et al. Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin Cancer Res. 2014;20:3507–3520. doi: 10.1158/1078-0432.CCR-13-2769. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 47.Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–2557. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.