

Abstract
Donohue syndrome is a rare autosomal recessive condition caused by severe loss-of-function mutations in the insulin receptor (INSR) gene. The diagnosis is made on clinical, biochemical and genetic grounds. Mutations are found on chromosome 19p13.2, and code for mutations in the INSR gene. Treatment is challenging and often unsuccessful, and relies on maintaining normoglycaemia and avoiding fasting; in some patients, recombinant human insulin-like growth factor (rhIGF-1) has been trialled. The prognosis is poor, with most babies dying in infancy. Ethically, it is important to consider the benefit versus burden of treatment, the quality of life of the surviving patient and the parents’ wishes, when making decisions regarding withholding or withdrawing care.
Background
Donohue syndrome1 is a rare (<1/1 000 000) autosomal recessive condition caused by severe loss-of-function mutations in the insulin receptor (INSR) gene.2 The diagnosis is based on a triad of clinical, biochemical and genetic findings. Clinically, common findings include intrauterine growth retardation and growth faltering in the newborn period. Affected infants are dysmorphic, with large and low-set ears, prominent eyes, broad nasal tip, flared nares, thick lips and gingival hyperplasia. They also have reduced subcutaneous fat, a distended abdomen and enlarged external genitalia and nipples.
Common biochemical findings in infants with Donohue syndrome include fasting hypoglycaemia and hyperinsulinism with post-prandial hyperglycaemia, due to impaired insulin sensitivity. Fasting C-peptide is also increased. Genetic mutations for Donohue syndrome are located on chromosome 19p. Donohue syndrome has a poor prognosis, and limited data suggest that many affected babies die in infancy and the majority die before reaching 2 years of age,2 usually related to infections.
Our case of Donohue syndrome highlights the clinical features of this rare disorder, while also examining the ethical challenges in achieving the appropriate level of care and treatment for a patient with this life-limiting condition.
Case presentation
Our case describes a male infant, delivered at 35 weeks gestation due to fetal distress and polyhydramnios, with very low birth weight (1.5 kg). Dysmorphic features and significant abdominal distension were noted immediately after delivery (figures 1 and 2). Extensive investigations performed soon after birth failed to yield a diagnosis. However, after several weeks, persistent hyperglycaemia in the context of his dysmorphic features, suggested a possible diagnosis of Donohue syndrome.
Figure 1.
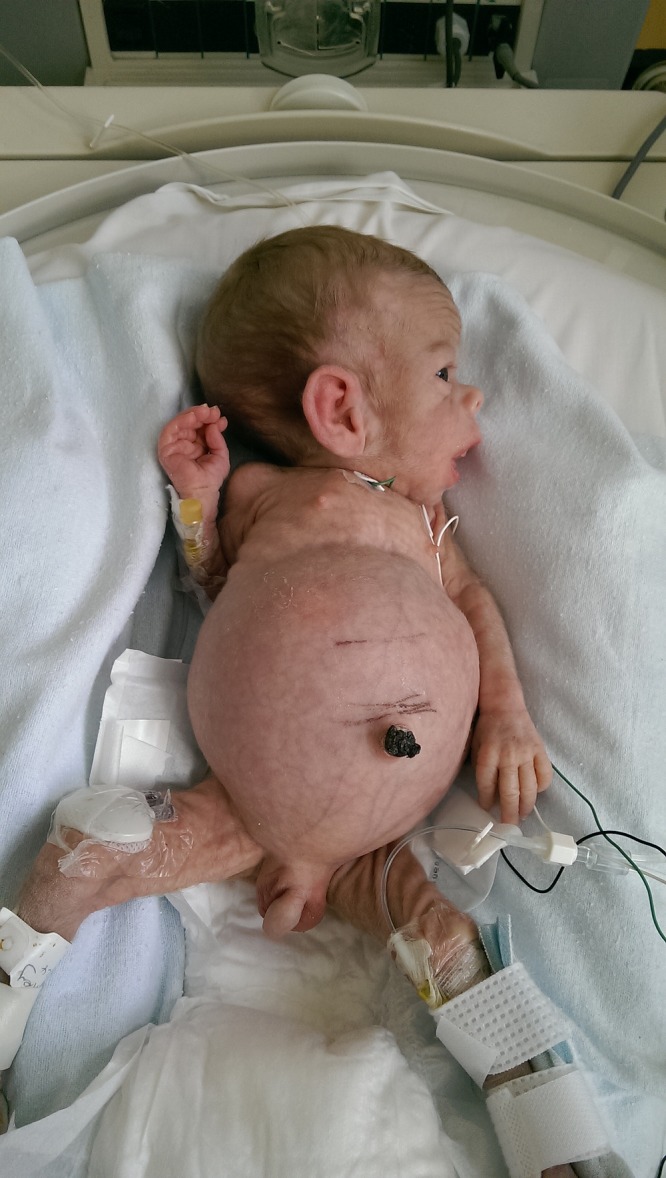
Clinical features.
Figure 2.

Clinical features.
Continuous intravenous insulin treatment was started for sustained hyperglycaemia (blood sugars up to 20 mmol/L) with glycosuria and polyuria. Wide fluctuations in blood sugars were observed, notably hypoglycaemia, when feeds were discontinued, and significant hyperglycaemia in the context of stress and sepsis. Ketone bodies were never identified. Insulin requirements were variable but peaked at 0.85 units/kg/h to maintain blood glucose in the 4–10 mmol/L range, suggesting impaired insulin sensitivity. An extensively hydrolysed casein formula at total fluid intake of 150 mL/kg/day, which appeared to be the maximum volume he could tolerate, via continuous nasogastric tube feeds, was delivered for nutrition. Despite the above regimen, our infant's weight plateaued at 1.6 kg. When intravenous access became difficult to maintain, a continuous subcutaneous insulin infusion (CSII) with sensor-augmented pump (SAP) was started to achieve ongoing normoglycaemia for treatment of permanent neonatal diabetes mellitus. CSII with SAP was chosen due to ongoing wide fluctuations in blood sugars on intravenous insulin. Insertion of these devices was challenging due to the baby's small size and lack of adiposity. Subsequently, at the request of his parents, SAP was discontinued and replaced with intermittent subcutaneous glucose monitoring. Over time, basal insulin requirements stabilised at 0.1 IU/h. Hypoglycaemia was only observed when continuous feeds were discontinued and several times when abdominal distention deteriorated; it is unclear how the two were related but perhaps there was malabsorption present simultaneously with hypoglycaemia.
Investigations
As outlined above, our patient was started on regular insulin therapy, unfortunately, an insulin level was not taken prior to treatment, however, the C-peptide level was elevated at 156.84 µg/L (reference range is 0.8–5.2 if fasting for ≥12 h, and 2–9 if post prandial). This increased C-peptide level can be interpreted as a surrogate marker of high insulin levels.
Genetic analysis performed for syndromic permanent neonatal diabetes mellitus identified a previously reported INSR missense mutation, p.A119V,3 and an INSR missense mutation, p.M1180K. The former and previously reported mutation causes a defect in the extracellular portion of the INSR, leading to decreased function. The latter novel mutation, which our patient had, also led to a missense mutation of the INSR. These compound heterozygote mutations at exon 2 and exon 20 are likely to have caused loss of function in the INSR, which would be consistent with the clinical picture and a diagnosis of Donohue syndrome.
Treatment
Currently, there is a lack of evidence for optimal treatment for Donohue syndrome. Therapy involves optimising glucose control; continuous feeding may be beneficial. Oral agents such as metformin and thiazolidinediones are not of benefit in the long term, as insulin replacement, eventually with very high doses, is necessary once diabetes has supervened.4 Some studies have shown positive results using recombinant human insulin-like growth factor (rhIGF-1); indeed, the recent literature has shown that continuous rhIGF-1 via an insulin pump may be a viable option for lowering glycated haemoglobin and improving weight gain.5
We decided to treat with insulin due to the persistent hyperglycaemia with accompanying glycosuria, polyuria and faltering growth. The insulin improved the baby's glucose level, on both intermittent subcutaneous glucose checks as well as on continuous glucose monitoring system. We set a target of 4–10 and adjusted insulin to maintain sugar within these targets.
Following lengthy discussions with his parents, they agreed to CSII therapy, and described that they were comfortable with this treatment, due to insulin use in various family members. The decision on whether or not to trial rhIGF-1 was deferred, pending evaluation of CSII therapy and further discussions with the parents. Ultimately, when the decision was taken to allow palliative care, rhIGF-1 and other extraordinary treatment options were not pursued.
Discussion
With confirmation of the diagnosis of a life-limiting condition, we were faced with significant ethical dilemmas regarding the optimum management plan for this infant and his family. Ongoing discussions between the multidisciplinary team (MDT) and the family were supported, to identify the optimum level of care for this infant, seeking to balance benefits of more therapy with the risks and consequences of more therapy.
Consideration of the family's wishes was a priority. Before genetic confirmation of Donohue syndrome was established, parental written consent was obtained that the infant was not for resuscitation in the event of a cardiopulmonary collapse. After the genetic diagnosis, and based on ongoing clinical status, his level of care and interventions were gradually de-escalated. Intravenous access was removed and no further blood tests (apart from subcutaneous) or intravenous antibiotics were planned. Treatment was moved towards comfort care and the palliative care team was introduced to the family.
Several important palliative care issues are noteworthy. First, central to the ethical argument about withholding versus withdrawing care, is to assess whether the patient is deemed to have a prognosis with the so-called ‘no chance’ or ‘no purpose’, or whether the patient falls into the ‘unbearable situation’.6 The ‘no chance’ situation refers to a child with such serious illness that life-sustaining treatment simply delays death without significant relief from suffering. The ‘no purpose’ situation refers to a patient who may be able to survive with treatment, but the degree of physical or mental impairment will be so significant that it is unreasonable to expect them to bear it. The ‘unbearable situation’ refers to a patient or their family who feels that, when faced with a progressive or irreversible disease, further treatment is more than can be tolerated. Our MDT discussions suggested that our patient probably met criteria for all three of these categories, given the extremely poor prognosis, treatment that would require repeated blood sampling and subcutaneous insulin administration, and, importantly, the parent's wishes to minimise pain and distress. It was on this basis we decided to evoke a palliative care plan. Furthermore, the MDT was mindful of the benefit versus burden of any treatment, and tried to emphasise achieving good quality of life for the patient and his family. Thus several questions were posed, including the following. Would the benefit of the insulin outweigh the burden? And, if so, what was the optimal method to deliver insulin to meet this particular baby's needs? As his prognosis was so poor, would treating high blood sugars improve his quality of life in a meaningful way? Are the repeated blood sugars and CSII site changes painful? Will CSII insertion become a problem due to his lipoatrophy? Perhaps, most importantly, we tried to rationalise whether any and all treatments were prolonging his life or prolonging his death?
Outcome and follow-up
Ultimately, the family played a central role in establishing the optimum level of care and treatment plan for their child. The MDT tried to share all information available to us, on the diagnosis and the prognosis, with the family, and tried to come to an agreed decision for the benefit of the patient. The family expressed a wish to spend as much quality time with their child as possible, while also minimising his pain or distress. After a detailed consultative process, palliative care in the patient's home was the aim of treatment. To achieve this, he was discharged on CSII therapy, as his mother expressed concerns that he would become symptomatic or uncomfortable without insulin. We planned for once daily blood sugar checks (more in the event of appearing unwell or not tolerating continuous feeds) and CSII site change every 3 days. We felt this plan respected his family's wishes while minimising distress related to treatment. Ongoing follow-up with the local palliative care team, public health nurse, general practitioner and paediatrician was implemented, our patient died at home at 4 months of age, following a short respiratory illness. In retrospect, his mother described to the MDT that, in her opinion, and in her specific family's case, the CSII allowed her to avoid insulin injections and also helped to reassure her that her son did not have symptoms of hypoglycaemia or hyperglycaemia, and that the CSII helped to improve her and her son's quality of life.
Learning points.
The diagnosis of Donohue syndrome is made on clinical, biochemical and genetic grounds. Mutations are found on chromosome 19p13.2, and code for mutations in the insulin receptor gene.
Treatment is challenging and often unsuccessful,7 and relies on maintaining normoglycaemia and avoiding fasting; in some patients, recombinant human insulin-like growth factor 1 has been trialled. The prognosis is poor, with most babies dying in infancy.
Strongly consider neonatal diabetes mellitus (NDM) screening in any child presenting with diabetes mellitus at <6 months of age. There are some children who present between 6 and 9 months and fewer who present from 9 to 12 months of age, who will also have NDM on screening.8
Ethically, it is important to consider the benefit versus burden of treatment, the quality of life of any patient and the parents’ wishes, when making decisions regarding withholding or withdrawing care.
Acknowledgments
The authors would like to acknowledge Staff nurse Carmel Collins, who played a key role in helping us arrive at the correct diagnosis. They also thank Hilary Noonan and Dr Mary Devins for their significant contribution from a palliative care perspective.
Footnotes
Contributors: DH wrote the first draft and performed the final edit of the manuscript. All the authors contributed to the planning and content of this work.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Donohue WL, Uchida IA. Leprechaunism: a euphuism for a rare familial disorder. J Pediat 1954;45:505–19. 10.1016/S0022-3476(54)80113-2 [DOI] [PubMed] [Google Scholar]
- 2.Falik Zaccai TC, Kalfon L, Klar A et al. . Two novel mutations identified in familial cases with Donohue syndrome. Mol Genet Genomic Med 2014;2:64–72. 10.1002/mgg3.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longo N, Wang Y, Smith SA et al. . Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet 2002;11:1465–75. 10.1093/hmg/11.12.1465 [DOI] [PubMed] [Google Scholar]
- 4.Semple RK, Williams RM, Dunger DB. CLINICAL QUESTION: What is the best strategy for patients with severe insulin resistance? Clin Endocrinol (Oxf) 2010;73:286–90. 10.1111/j.1365-2265.2010.03810.x [DOI] [PubMed] [Google Scholar]
- 5.Weber DR, Stanescu DE, Semple R et al. . Continuous subcutaneous IGF-1 therapy via insulin pump in a patient with Donohue syndrome. J Pediatr Endocrinol Metab 2014;27:1237–41. 10.1515/jpem-2013-0402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.RCPCH. Withholding or withdrawing life sustaining treatment in children. 2nd edn 2004:10–11. [Google Scholar]
- 7.McDonald A, Williams RM, Regan FM et al. . IGF-I treatment of insulin resistance. Euro J Endocrinol 2007;157(Suppl 1):S51–6. 10.1530/EJE-07-0271 [DOI] [PubMed] [Google Scholar]
- 8.Rubio-Cabezas O, Flanagan SE, Damhuis A et al. . KATP channel mutations in infants with permanent diabetes diagnosed after 6 months of life. Pediatr Diabetes 2012;13:322–5. 10.1111/j.1399-5448.2011.00824.x [DOI] [PubMed] [Google Scholar]