

Abstract
Background
Different apoptosis pathways activate caspase-3. In a study involving 27 patients with traumatic brain injury (TBI), higher caspase-3 levels were found in contusion brain tissue resected from non-survivors than from survivors. The objective of this study was to determine whether there is an association in TBI patients between serum caspase-3 levels (thus using an easier, quicker, less expensive and less invasive procedure) and mortality, in a larger series of patients.
Methods
We carried out a prospective, observational and multicenter study in six Spanish Hospital Intensive Care Units including 112 patients with severe TBI. All had Glasgow Coma Scale (GCS) scores lower than 9. Patients with an Injury Severity Score (ISS) in non-cranial aspects higher than 9 were excluded. Blood samples were collected on day 1 of TBI to measure serum caspas-3 levels. The endpoint was 30-day mortality.
Results
We found that non-surviving patients (n = 31) showed higher (p = 0.003) serum caspase-3 levels compared to survivors (n = 81). Kaplan-Meier survival analysis showed a higher risk of death in TBI patients with serum caspase-3 levels >0.20 ng/mL than in patients with lower concentrations (Hazard Ratio = 3.15; 95 % CI = 1.40 to 7.08; P < 0.001). Multiple logistic regression analysis showed that serum caspase-3 levels > 0.20 ng/mL were associated with mortality at 30 days in TBI patients controlling for Marshall CT classification, age and GCS (Odds ratio = 7.99; 95 % CI = 2.116 to 36.744; P = 0.001).
Conclusions
The association between serum caspase-3 levels and mortality in TBI patients was the major novel finding of our study.
Keywords: Caspase-3, Patients, Mortality, Injury, Brain
Background
Traumatic brain injury (TBI) is an important cause of death, disability, and health resource consumption [1]. Head trauma causes two types of injury in the neural tissue. One is the primary injury, which refers to the initial physical forces applied to the brain at the moment of impact. The other is the secondary injury, which develops over a period of hours or days later, involving neuroinflammatory response, free radical generation and apoptosis.
The apoptotic process is one in which cells are actively eliminated by a programmed pathway during morphogenesis, tissue remodeling, and resolution of the immune response; and it is increased in TBI [2–5]. Apoptotic cell death occurs mainly through two different pathways: the extrinsic or death receptor pathway (type I cells), and the intrinsic or mitochondrial pathway (type II cells). Both pathways, intrinsic and extrinsic, activate caspase-3 which leads to death cell.
Increased caspase-3 in brain tissues has been found in animal models of TBI [6–14]. However, caspase-3 has been scarcely explored in TBI patients, in small studies and only in brain tissue or cerebrospinal fluid (CSF) [15–19]. Higher caspase-3 levels in CSF have been found in TBI patients than in controls [15–17], and in brain tissue of TBI patients than in controls [18, 19]. In one study involving 27 TBI patients, higher caspase-3 levels were found in the brain tissue of non-surviving than in surviving TBI patients [19]. Thus the objective of this study was to determine whether there is an association between serum caspase-3 levels and mortality in TBI patients, (thus using an easier, quicker, less expensive and less invasive procedure compared with the determination in CSF or brain tissue of previous studies), in a larger series of patients.
Methods
Design and subjects
We carried out a prospective, observational and multicenter study in six Spanish Hospital Intensive Care Units including 112 patients with severe TBI. The study was approved by the Research Ethics Committees of the 6 participating hospitals, which were: Hospital Universitario de Canarias (La Laguna), Hospital Universitario Nuestra Señora de Candelaria (Santa Cruz de Tenerife), Hospital Clínico Universitario de Valencia (Valencia), Hospital General de La Palma (La Palma), Hospital Universitario Dr. Negrín (Las Palmas de Gran Canaria), and Hospital Insular (Las Palmas de Gran Canaria). Written informed consent was obtained from the patients or their legal guardians.
The present study included patients with severe TBI, defined as Glasgow Coma Scale (GCS) lower than 9 points [20]. We excluded patients with Injury Severity Score (ISS) [21] in non-cranial aspects higher than 9 points, age less than 18 years, pregnancy, inflammatory or malignant disease.
Variables recorded
The following variables were recorded for each patient: brain lesion according to Marshall computed tomography (CT) classification [22], sex, age, Acute Physiology and Chronic Health Evaluation II (APACHE II) score [23], activated partial thromboplastin time (aPTT), bilirubin, cerebral perfusion pressure (CPP), creatinine, fibrinogen, GCS, glycemia, hemoglobin, ICP, international normalized ratio (INR), ISS, lactic acid, leukocytes, pressure of arterial oxygen (PaO2), fraction of inspired oxygen (FI02), platelets, sodium, and temperature. The endpoint of the study was 30-day mortality.
Determinations of caspase-3 serum levels
Blood samples were collected on day 1 of TBI (within the first 4 h after TBI) in tubes with separator gel. After coagulation during 10 min at room temperature, serum was obtained by centrifugation at 1000 g for 15 min. The samples were aliquoted and frozen at −80 °C until determination. All determinations were performed by laboratory technicians blinded to all clinical data. Assays were performed at the Laboratory Department of the Hospital Universitario de Canarias (La Laguna, Tenerife, Spain).
Caspase-3 levels were measured in serum by solid-phase enzyme-linked immunosorbent assay (ELISA) quantitative sandwich using Human Caspase-3 Elisa BlueGene Biotech® (Shanghai, China). The intra- and inter-assay coefficients of variation (CV) were <5.6 % and <7.9 % respectively. The detection limit for the assay was 0.1 ng/mL.
Statistical methods
Quantitative variables are reported as medians and interquartile ranges, and were compared with Wilcoxon-Mann–Whitney test. Qualitative variables are reported as frequencies and percentages and compared using Chi-squared test.
We used receiver operating characteristic analysis to determine the goodness-of-fit of serum caspase-3 levels to predict 30-day mortality. We carried out a Kaplan-Meier analysis to compare 30-day survival according to serum caspase-3 levels lower/higher than 0.20 ng/mL.
We constructed predictive Cox regression models of survival after TBI. The variables included were: serum caspase-3 levels > 0.20 ng/mL, Marshall CT classification, GCS and age as independent variables. To include the variable CT classification in the regression analysis, it was recoded according to the risk of death observed in the bivariate analysis as high risk of death (CT types 3, 4 and 6) and low risk of death (CT types 2 and 5). The dependent variable was time to death. We included −2 log likelihood ratios to compare the models. We used Hazard ratio and 95 % confidence interval (CI) to estimate the clinical impact of the predictor variables.
SPSS 17.0 (SPSS Inc., Chicago, IL, USA) and LogXact 4.1, (Cytel Co., Cambridge, MA) was used to perform statistical analyses. All P values lower 0.05 were considered statistically significant
Results
At 30 days after TBI, 31 of the 112 (27.7 %) patients had died. We observed a lower proportion of female patients (p = 0.02) among surviving than non-surviving TBI patients, and different CT findings (p = 0.003) (Table 1). Surviving TBI patients showed higher GCS (p < 0.001), lower age (p < 0.001), and lower APACHE-II score (p < 0.001) than non-survivors. In addition, surviving patients showed lower serum caspase-3 levels than non-survivors (p = 0.003) (Table 2).
Table 1.
Comparison of computed tomography findings and gender between surviving and non-surviving patients with traumatic brain injury
| Survivors (n = 81) | Non-survivors (n = 31) | P-value | |
|---|---|---|---|
| Marshall CT classification - n (%) | 0.003 | ||
| Type 6 | 5 (6.2) | 8 (25.8) | |
| Type 5 | 26 (32.1) | 5 (16.1) | |
| Type 4 | 12 (14.8) | 9 (29.0) | |
| Type 3 | 14 (17.3) | 6 (19.4) | |
| Type 2 | 24 (29.6) | 3 (9.7) | |
| Type 1 | 0 | 0 | |
| CT findings with high risk of death (types 3,4,6) - n (%) with classification - n (%) | 31 (38.3) | 23 (74.2) | 0.001 |
| Female gender – n (%) | 13 (16.0) | 12 (38.7) | 0.02 |
Data are shown as number and percentage; CT Computed tomography
Table 2.
Comparison of clinical and biochemical characteristics between surviving and non-surviving patients with traumatic brain injury
| Survivors (n = 81) | Non-survivors (n = 31) | P-value | |
|---|---|---|---|
| Age (years) | 46 (27–60) | 63 (53–75) | <0.001 |
| APACHE-II score | 18 (15–22) | 26 (24–29) | <0.001 |
| aPTT (seconds) | 28 (25–31) | 29 (25–36) | 0.86 |
| Bilirubin (mg/dl) | 0.50 (0.40–0.80) | 0.70 (0.58–0.95) | 0.045 |
| Caspase-3 (ng/ml) | 0.11 (0.10–0.18) | 0.23 (0.10–0.46) | 0.003 |
| CPP (mmHg) | 68 (57–70) | 61 (54–69) | 0.48 |
| Creatinine (mg/dl) | 0.80 (0.63–1.00) | 0.80 (0.70–1.10) | 0.44 |
| Fibrinogen (mg/dl) | 366 (283–448) | 360 (269–520) | 0.32 |
| Glasgow Coma Scale score | 7 (5–8) | 3 (3–6) | <0.00171 |
| Glycemia (g/dL) | 139 (122–167) | 160 (134–189) | 0.08 |
| Hemoglobin (g/dL) | 11.4 (10.2–13.0) | 11.9 (9.8–13.1) | 0.87 |
| ICP (mmHg) | 15 (14–20) | 25 (13–34) | 0.28 |
| INR | 1.11 (1.00–1.21) | 1.12 (1.03–1.40) | 0.29 |
| ISS | 25 (25–29) | 25 (25–29) | 0.80 |
| Lactic acid (mmol/L) | 1.70 (1.10–2.50) | 2.40 (1.30–4.60) | 0.06 |
| Leukocytes *103/mm3 | 14.5 (10.3–19.0) | 16.3 (9.8–22.7) | 0.46 |
| PaO2 (mmHg) | 148 (110–203) | 133 (98–180) | 0.33 |
| PaO2/FI02 ratio | 336 (240–400) | 274 (173–393) | 0.11 |
| Platelets *103/mm3 | 184 (134–244) | 180 (125–237) | 0.52 |
| Sodium (mEq/L) | 140 (138–143) | 142 (138–148) | 0.19 |
| Temperature (°C) | 37.0 (36.0–37.3) | 36.0 (35.0–37.0) | 0.12 |
Data are shown as median (percentile 25th–75th); APACHE II Acute Physiology and Chronic Health Evaluation, aPTT activated partial thromboplastin time, CPP cerebral perfusion pressure, ICP intracranial pressure, INR international normalized ratio, ISS Injury Severity Score, PaO 2 pressure of arterial oxygen, FIO 2 fraction of inspired oxygen
On receiver operating characteristic analysis, the area under the curve of serum caspase-3 levels to predict morality at 30 days in TBI patients was 0.68 (95 % CI = 0.56–0.80; P = 0.003).
Kaplan-Meier survival analysis showed a higher risk of death in TBI patients with serum caspase-3 levels >0.20 ng/mL than in patients with lower concentrations (Hazard Ratio = 3.15; 95 % CI = 1.40 to 7.08; P < 0.001) (Fig. 1).
Fig. 1.
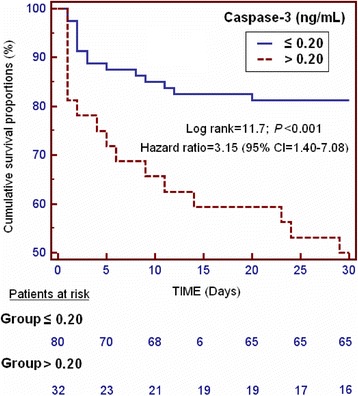
Survival curves at 30 days using serum caspase-3 levels higher or lower than 0.20 ng/mL
Cox regression analysis showed that serum caspase-3 levels >0.20 ng/mL were associated with mortality at 30 days in TBI patients after controlling for Marshall CT classification, age and GCS (Hazard ratio = 2.956; 95 % CI = 1.365 to 6.403; P = 0.006) (Table 3).
Table 3.
Cox regression analysis to predict mortality
| −2 log likelihood ratio | Hazard Ratio | 95 % Confidence Interval | P-value | |
|---|---|---|---|---|
| Model 1: | 274.431 | 3.163 | 1.562–6.405 | 0.001 |
| Serum caspase-3 > 0.20 ng/mL | ||||
| Model 2: | 263.541 | |||
| Serum caspase-3 > 0.20 ng/mL | 2.916 | 1.438–5.917 | 0.003 | |
| CT findings (high vs low risk of death) | 3.538 | 1.577–7.940 | 0.002 | |
| Model 3: | 234.391 | |||
| Serum caspase-3 > 0.20 ng/mL | 2.943 | 1.396–6.203 | 0.005 | |
| Glasgow Coma Scale | 0.703 | 0.586–0.845 | <0.001 | |
| Model 4: | 240.152 | |||
| Serum caspase-3 > 0.20 ng/mL | 3.349 | 1.605–6.990 | 0.001 | |
| Age (years) | 1.039 | 1.017–1.061 | 0.001 | |
| Model 5: | 209.615 | |||
| Serum caspase-3 > 0.20 ng/mL | 2.956 | 1.365–6.403 | 0.006 | |
| CT findings (high vs low risk of death) | 4.289 | 1.836–10.016 | 0.001 | |
| Glasgow Coma Scale | 0.694 | 0.567–0.849 | <0.001 | |
| Age (years) | 1.036 | 1.015–1.058 | 0.001 |
CT Computed tomography
Discussion
The novel findings of our study were that non-surviving TBI patients showed higher serum caspase-3 levels than survivors, that high serum caspase-3 levels were associated with higher mortality, and that serum caspase-3 levels could be used as a biomarker to predict mortality in TBI patients.
Previously, higher caspase-3 levels were found in the CSF of TBI patients than in controls [15–17], and in brain tissue of TBI patients than in controls [18, 19]. In our study we found higher serum caspase-3 levels in non-surviving than in surviving TBI patients. These results are in consonance with those of a study by Nathoo et al. [19] involving 27 TBI patients, which showed higher caspase-3 levels in the brain tissue of non-survivors than survivors. The new aspect of our study was that caspase-3 levels were measured for the first time in serum; thus using an easier, quicker, less expensive and less invasive procedure compared with the determination in CSF or brain tissue of previous studies. The major novel finding of our study, according to the results of Cox regression analysis, was that serum caspase-3 levels >0.20 ng/mL were associated with 30-day mortality in TBI patients.
The findings of our study suggest that apoptosis in patients with severe TBI may play an important role in prognosis. Apoptotic cell death occurs primarily through three different pathways: the extrinsic or death receptor pathway (type I cells), the intrinsic or mitochondrial pathway (type II cells), and the endoplasmic reticulum pathway [2–5]). In type I cells, the activation of a surface death receptor of tumor necrosis factor receptor superfamily (TNFRSF) by its cognate death ligand (TNFSF) initiates apoptosis, then a death signal is created and cleaves pro-caspase-8 in active caspase-8, which activates caspase-3. In type II cells, apoptosis could be initiated by cytokines such as interleukin (IL)-1 and IL-6, and oxygen free radicals; in this pathway the mitochondria release cytochrome c which activates caspase 3. Both apoptotic pathways, intrinsic and extrinsic, activate caspase-3 and that leads to cell death. Caspase-3 cleaved DNA fragmentation factor subunit alpha (DFFA), also known as Inhibitor of caspase-activated DNase (ICAD), and triggers DNA fragmentation during apoptosis [24].
In rat models, the administration of caspase-3 inhibitors have reduced caspase-3 activity and apoptosis in brain tissues [9–14]. Thus, from a therapeutic perspective, the use of modulators of apoptotic activity by inhibition of caspase-3 activity could be new class of drugs for the treatment of TBI.
Our study has certain limitations. First, the determination of circulating caspase-3 levels during follow-up was not performed and could be interesting. Second, we did not determine serum caspase-3 levels in healthy controls; however, the objective of our study was not to determine whether there is an increase of serum caspase-3 levels in TBI patients, but rather whether there is an association between serum caspase-3 levels and 30-day mortality in TBI patients. Third, the inclusion and exclusion criteria of our study were fairly restrictive, which hinders extrapolation of the findings to the general TBI population. However, we excluded patients with ISS in non-cranial aspects higher than 9 or inflammatory disease to avoid the influence of inflammation on serum caspase-3 levels; and we excluded patients with malignant disease to avoid the influence of therapeutic effort limitation on prognosis. Four, blood samples were collected on the day of TBI (within the first 4 h after TBI); however, the exact moment of blood sampling was not reported and the time between the moment of trauma and blood sampling may be different among patients. Five, the findings of our study suggest that there is an association between serum caspase-3 levels and mortality in TBI patients and that serum caspase-3 levels could be used as a prognostic biomarker of early mortality; however, these findings do not imply a causal relationship. Thus, additional studies are needed to confirm the results of our study
Conclusions
The association between serum caspase-3 levels and 30-day mortality in TBI patients was the major novel finding of our study.
Abbreviations
- TBI
Traumatic brain injury
- GCS
Glasgow Coma Scale
- ISS
Injury Severity Score
- PaO2
Pressure of arterial oxygen/fraction inspired oxygen
- FIO2
Pressure of arterial oxygen/fraction inspired oxygen
- aPTT
activated partial thromboplastin time
- APACHE II
Acute Physiology and Chronic Health Evaluation
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: LL. Acquired the data: LL, MMM, MA, LR, JSV, MRR. Serum caspase-3 analysis: JMBL. Analized the data: LL, AJ. Wrote the paper: LL. All authors revised the manuscript critically for important intellectual content and made the final approval of the version to be published.
Contributor Information
Leonardo Lorente, Email: lorentemartin@msn.com.
María M. Martín, Email: mar.martinvelasco@gmail.com
Mónica Argueso, Email: moni_begasa@hotmail.com.
Luis Ramos, Email: lramosgomez@gmail.com.
Jordi Solé-Violán, Email: jsolvio@gobiernodecanarias.org.
Marta Riaño-Ruiz, Email: mriarui@gobiernodecanarias.org.
Alejandro Jiménez, Email: ajimenezsosa@gmail.com.
Juan M. Borreguero-León, Email: jmborreguero@hotmail.com
References
- 1.Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24:S1–106. doi: 10.1089/neu.2006.0209. [DOI] [PubMed] [Google Scholar]
- 2.Cavallucci V, D’Amelio M. Matter of life and death: the pharmacological approaches targeting apoptosis in brain diseases. Curr Pharm Des. 2011;17:215–29. doi: 10.2174/138161211795049705. [DOI] [PubMed] [Google Scholar]
- 3.Wang K, Liu B, Ma J. Research progress in traumatic brain penumbra. Chin Med J (Engl) 2014;127:1964–8. [PubMed] [Google Scholar]
- 4.Rovegno M, Soto PA, Sáez JC, von Bernhardi R. Biological mechanisms involved in the spread of traumatic brain damage. Med Intensiva. 2012;36:37–44. doi: 10.1016/j.medin.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Kunz A, Dirnagl U, Mergenthaler P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract Res Clin Anaesthesiol. 2010;24:495–509. doi: 10.1016/j.bpa.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Jia F, Mao Q, Liang YM, Jiang JY. Effect of post-traumatic mild hypothermia on hippocampal cell death after traumatic brain injury in rats. J Neurotrauma. 2009;26:243–52. doi: 10.1089/neu.2008.0670. [DOI] [PubMed] [Google Scholar]
- 7.Eberspächer E, Heimann K, Hollweck R, Werner C, Schneider G, Engelhard K. The effect of electroencephalogram-targeted high- and low-dose propofol infusion on histopathological damage after traumatic brain injury in the rat. Anesth Analg. 2006;103:1527–33. doi: 10.1213/01.ane.0000247803.30582.2d. [DOI] [PubMed] [Google Scholar]
- 8.Shah SA, Prough DS, Garcia JM, DeWitt DS, Hellmich HL. Molecular correlates of age-specific responses to traumatic brain injury in mice. Exp Gerontol. 2006;41:1201–5. doi: 10.1016/j.exger.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Saykally JN, Rachmany L, Hatic H, Shaer A, Rubovitch V, Pick CG, et al. The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience. 2012;223:305–14. doi: 10.1016/j.neuroscience.2012.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, et al. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol. 2006;197:437–50. doi: 10.1016/j.expneurol.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Soustiel JF, Palzur E, Nevo O, Thaler I, Vlodavsky E. Neuroprotective anti-apoptosis effect of estrogens in traumatic brain injury. J Neurotrauma. 2005;22:345–52. doi: 10.1089/neu.2005.22.345. [DOI] [PubMed] [Google Scholar]
- 12.Clausen F, Lundqvist H, Ekmark S, Lewén A, Ebendal T, Hillered L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J Neurotrauma. 2004;21:1168–82. doi: 10.1089/neu.2004.21.1168. [DOI] [PubMed] [Google Scholar]
- 13.Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740–53. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–9. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 15.Uzan M, Erman H, Tanriverdi T, Sanus GZ, Kafadar A, Uzun H. Evaluation of apoptosis in cerebrospinal fluid of patients with severe head injury. Acta Neurochir (Wien) 2006;148:1157–64. doi: 10.1007/s00701-006-0887-1. [DOI] [PubMed] [Google Scholar]
- 16.Härter L, Keel M, Hentze H, Leist M, Ertel W. Caspase-3 activity is present in cerebrospinal fluid from patients with traumatic brain injury. J Neuroimmunol. 2001;121:76–8. doi: 10.1016/S0165-5728(01)00409-X. [DOI] [PubMed] [Google Scholar]
- 17.Hentze H, Schwoebel F, Lund S, Keel M, Ertel W, Wendel A, et al. In vivo and in vitro evidence for extracellular caspase activity released from apoptotic cells. Biochem Biophys Res Commun. 2001;283:1111–7. doi: 10.1006/bbrc.2001.4918. [DOI] [PubMed] [Google Scholar]
- 18.Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–21. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- 19.Nathoo N, Narotam PK, Agrawal DK, Connolly CA, van Dellen JR, Barnett GH, et al. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J Neurosurg. 2004;101:233–40. doi: 10.3171/jns.2004.101.2.0233. [DOI] [PubMed] [Google Scholar]
- 20.Teasdale G, Jennett B. Assessement of coma and impaired conciousness. A practical scale. Lancet. 1974;2:81–4. doi: 10.1016/S0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 21.Baker SP, O’Neill B, Haddon W, Jr, Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J Trauma. 1974;14:187–96. doi: 10.1097/00005373-197403000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Marshall LF, Marshall SB, Klauber MR, Van Berkum CM, Eisenberg H, Jane JA, et al. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(Suppl 1):S287–92. [PubMed] [Google Scholar]
- 23.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–29. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–84. doi: 10.1016/S0092-8674(00)80197-X. [DOI] [PubMed] [Google Scholar]