

Abstract
Administration of intravenous immunoglobulin (IVIg) is a recognized safe and efficient immunomodulation therapy for many autoimmune diseases. Anti-idiotypic antibody binding to pathogenic autoantibodies was proposed as one of the mechanisms attributed to the protective activity of IVIg in autoimmunity. The aim of this study was to fractionate the anti-anti-citrullinated protein anti-idiotypic-antibodies (anti-ACPA) from an IVIg preparation and to test it as a treatment for collagen-induced arthritis in mice. IVIg was loaded onto an ACPA column. The eluted fraction was defined as ACPA-specific-IVIg (ACPA-sIVIg). Collagen-induced-arthritis (CIA) was induced in mice. Mice were treated weekly with ACPA-sIVIg, low-dose-IVIg, high-dose-IVIg and phosphate-buffered saline (PBS). Sera-ACPA titres, anti-collagen anitbodies and cytokine levels were analysed by enzyme-linked immunosorbent assay (ELISA); antibody-forming-cell activity by enzyme-linked imunospot (ELISPOT) assay; and expansion of regulatory T cell (Treg) population by fluorescence activated cell sorter (FACS). ACPA-sIVIg inhibited ACPA binding to citrullinated-peptides (CCP) in vitro 100 times more efficiently than the IVIg compound. ACPA-sIVIg was significantly more effective than the IVIg-preparation in attenuating the development of collagen-induced arthritis. Splenocytes from CIA mice treated with ACPA-sIVIg reduced the ACPA and anti-collagen-antibody titres, including the number of anti-collagen and ACPA antibody-forming cells. In parallel, splenocytes from ACPA-sIVIg treated mice secreted higher levels of anti-inflammatory cytokines and lower proinflammatory cytokines. The ACPA-sIVIg inhibitory potential was accompanied with expansion of the Treg population. Low-dose IVIg did not affect the humoral and cellular response in the CIA mice in comparison to the PBS-treated mice. Based on our results, IVIg may be considered as a safe compound for treating patients with rheumatoid arthritis by neutralizing pathogenic autoantibodies, reducing proinflammatory cytokines and expanding the Treg population.
Keywords: ACPA, autoimmunity, collagen-induced arthritis, IVIg, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is an autoimmune rheumatic disease that primarily affects the joints and synovial tissue, resulting in inflammation and destruction. Anti-citrullinated protein antibodies (ACPA) are detected in approximately 90% of RA patients and are involved in the pathogenesis of the disease 1–5. During the process of inflammation, arginine amino acid residues are converted enzymatically into citrulline in proteins such as collagen, enolase, vimentin, fibrin and fibrinogen, a process termed citrullination. ACPA are pathogenic autoantibodies that can initiate and enhance arthritis in murine models 1–4. The mechanism by which ACPA promotes inflammation was proposed to be related to the ACPA activating complement and diverse cells of the immune system. The cell perturbation was attributed to ACPA binding to the Fc-receptor, F(ab)2 or to cross-linking between the Fc-receptor and B cell receptor (BCR) by ACPA. This scenario involves the complement system, causing secretion of proinflammatory cytokines such as tumour necrosis factor (TNF)-α, recruitment of inflammatory-related cell subsets and formation of neutrophil extracellular traps (NETs) (NETosis) leading to inflammation of the joints 1–5. The involvement of ACPA in the inflammatory process indicates the need for therapies aimed at blocking the activity of pathogenic autoantibodies.
Intravenous immunoglobulin (IVIg) therapy has been shown to be beneficial in various autoimmune diseases 6–8. IVIg has a therapeutic effect in RA in human and animal models such as collagen-induced arthritis (CIA) 9–12. IVIg immunomodulate the humoral and cellular responses of the immune network through the Fab and Fc portions of the immunoglobulins 7–16. Among the diverse pathways of IVIg activities in autoimmunity, anti-idiotypic activity has gained a great deal of attention 13,14,16. Previously, we and others have shown the immunomodulating effects of specific anti-idiotypic IVIg in animal models of autoimmune diseases such as lupus, anti-phospholipid syndrome, myasthenia gravis, vasculitis and pemphigus vulgaris 17–21. In the current study, we examined the efficacy of anti-ACPA-specific IVIg (anti-Id) in the treatment of mice with CIA.
Methods
Fractionation of ACPA-specific IVIg (ACPA-sIVIg)
IVIg was supplied by Laboratoire Français des Biotechnologies, LFB-Tegeline, France. The high-dose concentration of IVIg used in the study was 3 mg/ml; the low-dose IVIg concentration was 30 μg/ml.
Stage I
For purification of ACPA, total IgG was affinity purified on a Protein-G column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Four ml of patient serum diluted five times with phosphate-buffered saline (PBS) was allowed to pass through the column for 4 h at room temperature. The procedure was repeated for 12 patients. The column was then washed with PBS and bound antibodies were eluted with 0·2 M glycin-HCl pH 2·2, neutralized to pH 7 and dialysed against PBS.
ACPA from the total IgG was purified on a citrullinated peptide column constructed using a HiTrap column (GE Healthcare). The citrullinated peptides used in the study comprised the following 22,23:
Cit-fillagrin (cfc6)306SHQEST(Cit)G(Cit)SRGRSGRSGS324 –K-NH2;
Cit-collagen: 359A(Cit)GLTG(Cit)PGDA369-K-NH2;
Cit-vimentin 65SAVRA(Cit)SSVPGVR77-K-NH2;
Cit-β-fibrinogen 60(Cit)PAPPPISGGGY(Cit)A(Cit)74-K-NH2; abd
α-enolase 5KIHA(Cit)EIFDS(Cit)GNPTVE21-K-NH2.
HiTrap™ N-hydroxysuccinimide (NHS)-activated-Sepharose™ High Performance (GE Healthcare). The citrullinated peptides were dissolved in coupling buffer (0·2 M NaHCO3, 0·5 M NaCl, pH 8·3), 5 mg/10 ml. The isopropanol was washed out with 3 × 2 ml ice-cold 1 mM HCl. Two ml of coupling buffer was added, followed by the peptides solution into the column using a pump at for 4 h at room temperature. The non-bound peptides were washed and deactivated by 0·5 M ethanolamine 0·5 M NaCl, pH 8·3 buffer followed by 0·1 M acetate, 0·5 M NaCl, pH 4. The pH was adjusted with 0·05 M Na2HPO4, pH 7. Total fractionated IgG was loaded 2 mg/ml in PBS onto the peptides column. Following extensive washing, the bound ACPA was eluted with 0·2 M glycin-HCl, pH 2·5, neutralized immediately to pH 7 and dialysed with PBS.
Stage II
For the construction of the ACPA column, a mix of affinity-purified ACPA antibodies from 12 patients with RA were dialysed against coupling buffer (0·1 M NaHCO3, pH 8·3, containing 0·5 M NaCl) overnight at 4°C and bound covalently to CN.Br-activated Sepharose 4B (GE Healthcare), according to the manufacturer's instructions. The remaining active NH groups were blocked by incubation with 0·2 M glycine, pH 8·0, overnight at 4°C. Three washing cycles with coupling buffer, followed by 0·1 M acetate buffer, pH 4·0, were used to remove the excess of unbound ACPA. The washed column was equilibrated in sterile Tris-HCl buffer pH 7·4.
Stage III
For fractionation of IVIG anti-ACPA idiotypic antibodies–sIVIG, 500 mg of the commercial IVIG preparation (Omrix, Ness-Ziona, Israel) were dialysed against loading buffer (0·05 M Tris containing 0·5 M NaCl, pH 8·0), diluted in 50 ml of loading buffer, filtered through the 0·45 μm filter (Minisart, Sartorius AG, Göttingen, Germany) and passed through the ACPA column for 16 h at 4°C. Unbound material was washed out and the bound antibodies were eluted with 0·2 M glycine-HCl, pH 2·2, and neutralized immediately to pH 7.
Mice
Male DBA/1J mice, 6–7 weeks old, were purchased from Harlan Laboratories (Venray, the Netherlands). The mice were kept in individually ventilated cages. All experiments were approved and executed according to the Ethical Committee of the Israeli Ministry of Health, protocol number 787/12.
Induction of arthritis and treatment
Experimental arthritis was induced in male DBA/1J 6–7-week-old mice with bovine type II collagen (CII), which is a prototype model of RA and shares many clinical and histopathological similarities to human RA 12,24. Bovine type II collagen 100 μg/mouse (CII, Chondrex Redmond, WA, USA) in 1 : 1 emulsion with Mycobacterium tuberculosis H37RA in Freund's incomplete adjuvant (Difco Laboratories, Detroit MI, USA) were injected at the base of the tail. Booster injection was given on day 21. Mice were monitored for signs of arthritis three times a week by two blinded observers. Severity scores were derived as follows: 0 = normal, 1 = erythema, 2 = erythema plus swelling, 3 = extension/loss of function and total score = sum of four limbs. The treatment started at day 0 when the disease was induced, intraperitoneally, with either ACPA-sIVIg 30 μg/0·1ml PBS, low-dose IVIg (L-IVIg) 30 μg/0·1ml PBS or high-dose IVIg (H-IVIg) 3 mg/0·1ml in PBS or with PBS. The treatments were given weekly. The mice were killed at days 38–40.
Histological analysis of the joints
Joints from the mice were then embedded in paraffin. The pathology of the joints was identified by histological staining [haematoxylin and eosin (H&E)]. The evaluation was performed by a pathologist.
Screening for autoantibodies by enzyme-linked immunosorbent assay (ELISA)
The determination of circulating mouse ACPA and anti-collagen antibodies was performed as follows: human ACPA were detected using collagen-coated cell culture plates (CCP) (Quanta Lite CCP3 IgG ELISA; Inova Diagnostics, Inc., San Diego, CA, USA). Mouse autoantibodies were studied in sera from the different groups of mice. ELISA plates were coated with collagen-II or peptide-mix 10 μg/ml PBS overnight at 4°C. Following blocking with 3% bovine serum albumin (BSA), sera were applied at a 1 : 200 dilution for 2 h. The binding was probed by goat anti-mouse-IgG-alkaline-phosphatase, diluted 1 : 5000 with the appropriate substrate. Data were read by ELISA reader at 405 nm optical density (OD).
Antibody-forming cell activity
In order to clarify whether the lower sera titres of autoantibodies in the ACPA-sIVIg-treated group of mice resulted from fewer B cell-producing antibodies or the decreased ability of B cells to secrete antibodies, we affinity-purified B cells by negative selection using magnetic microbeads (MACS; Miltenyi Biotec, Bergisch Gladbach, Germany) and performed the antibody-producing cells assay in vitro using the enzyme-linked immunospot (ELISPOT) blue colour module ELISPOT assay (R&D Systems, Minneapolis, MN, USA). The assay was performed using a 96-well polyvinylidene difluoride (PVDF)-backed microplate, according to the manufacturer's protocol. Briefly, 30 µg/ml of citrullinated peptide mix or collagen type II in 100 µl dilution buffer was applied per well to the ELISPOT plate and incubated overnight at 4°C. Following washings and blocking with RPMI-1640 supplemented with 10% fetal calf serum (FCS) for 2 h, affinity-purified B cells were seeded at 105 cells/well and incubated for 24 h at 37°C in 5% CO2. Following washing of the cells, the spots of the secreted antibodies were probed with a second antibody goat-anti-mouse IgG conjugated to horseradish peroxidase (HRP) and a specific substrate. The spots were counted.
Cytokine secretion
Splenocytes from the different groups of mice were cultured 5 × 106 cells/well in 12-well plates coated with anti-CD3 1 μg/ml in an enriched medium (RPMI-1640 supplemented with 10% FCS, 1% penicillin/streptomycin, 2% glutamic acid and 10−5 M mercaptoethanol) at 37°C and 5% CO2. The splenocytes were incubated in the presence of L-IVIg 30 μg/ml, ACPA-sIVIg 30 μg/ml and H-IVIg 3 mg/ml. After 48 and 72 h, the supernatants were harvested for detection of TNF-α, interleukin (IL)-1β and IL-10. The cytokine levels were determined using the sandwich DuoSet ELISA kit (R&D systems), performed according to the manufacturer's instructions. For detection of transforming growth factor (TGF)-β, supernatants were added after activation of a latent TGF-β to immunoreactive TGF-β1. A biotinylated anti-human TGF-β1 antibody was added thereafter, and the assay was developed according to the manufacturer's instructions for the TGF-β kit (R&D Systems).
Flow cytometry analyses
The percentage of CD4+CD25+forkhead box protein 3 (FoxP3+) regulatory T cells (Tregs) in the splenocyte CD4+ T cell population from different groups of mice was analysed by fluorescence activated cell sorter (FACS). Fluorescein isothiocyanate (FITC) conjugated to anti-mouse-CD4, allophycocyanin (APC) conjugated to anti-mouse CD25 and phycoerythrn (PE) conjugated to anti-mouse/rat FoxP3 were used for the analyses (all from eBioscience, San Diego, CA, USA). After red blood cell lysis, splenocytes were incubated with anti-CD4 anti-CD25 monoclonal antibodies (mAbs) for 1·5 h at room temperature. Following washing with PBS, the cells were fixed and permeabilized with Fix/Perm buffer and permeabilization buffer (R&D Systems) overnight at 4°C. The next day, anti-FoxP3 was added for 1·5 h of incubation, washed and analysed by FACS. To visualize CD4+CD25+FoxP3+ cells, we gated on CD4+ T cells and analysed for the expression of CD25+FoxP3+. The total number of events was 50 000 per sample point.
Statistical analysis
Data are expressed as mean ± standard deviation (s.d.). Statistical analysis was performed with one-way analysis of variance (anova) followed by Student's t-test for comparison between groups. P-values < 0·05 were considered significant.
Results
Inhibition of ACPA binding to CCP by anti-idiotypic ACPA-sIVIg
In the first stage, we clarified whether the LFB-IVIg compound encompassed a subfraction of anti-idiotypic antibodies to ACPA. As illustrated in Fig. 1a, biotin-labelled IVIg at a concentration of 3 mg/ml showed significant ACPA binding to CCP, compared to the control IgG (P < 0.002). Whereas IVIg fractionated on an ACPA column, ACPA specific-IVIg (ACPA-sIVIg) bound ACPA at a concentration of 30 µg/ml (P < 0·002), 100 times stronger than the entire IVIg compound (Fig. 1b). IVIg and ACPA-sIVIg binding behaved in a dose-dependent manner, as illustrated in Fig. 1a,b.
Figure 1.
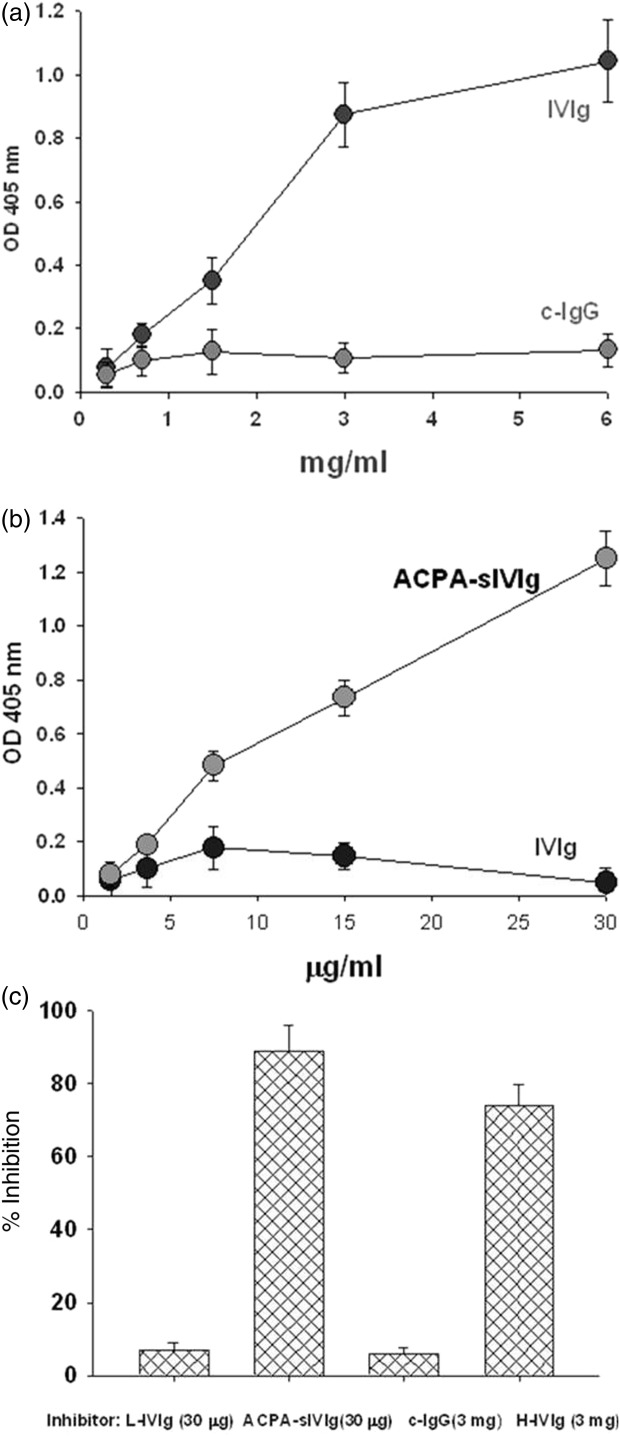
Characterization of anti-anti-citrullinated peptides antibodies (ACPA) intravenous immunoglobulin (IVIg): (a) anti-ACPA fraction in commercial IVIg. Laboratoire Français des Biotechnologies (LFB)-IVIg was tested for the presence of anti-ACPA IVIg on CCP enzyme-linked immunosorbent assay (ELISA) plate. The results are expressed in optical density (OD). At 405 nm, as mean ± standard deviation (s.d.) of three separate experiments. (b) Anti-idiotypical binding of ACPA-sIVIg. Binding of biotinylated-ACPA-specific IVIg (sIVIg) to ACPA on streptavidin–ACPA-bound ELISA plate. The results are expressed in OD at 405 nm, as mean ± s.d. of three separate experiments. (c) In-vitro efficacy of ACPA-sIVIg. IVIg fractions as inhibitors of ACPA binding to CCP. The results are expressed as percentage of inhibition of three separate experiments.
Inhibition experiments were conducted to confirm the stronger activity of ACPA-sIVIg. We studied the ability of ACPA-sIVIg to inhibit ACPA binding to CCP in compared to the entire IVIg compound. Data presented in Fig. 1c show that ACPA-sIVIg inhibited ACPA binding by 90% at a concentration of 30 µg/ml. IVIg at the same concentration inhibited the binding by only 4% (P < 0·001). High-dose IVIg (3 mg/ml) inhibited ACPA binding to CCP by 75% compared to the control IgG (P < 0·002). No significant difference was observed between sIVIg and IVIg in inhibiting the ACPA-CCP binding capacity (P > 0·05). These data indicate the advantage of sIVIG on the IVIg compound.
ACPA-sIVIg attenuated the development of CIA in mice
The clinical arthritis score was significantly lower in ACPA-sIVIg treated mice compared with groups treated with low-dose IVIg or PBS (P < 0·001) at day 38, as illustrated in Fig. 2a. ACPA-sIVIg treatment lowered the clinical score significantly more than H-IVIg at day 38 (P < 0·03). The histological analysis of the inflammation in the four joints is described in Fig. 2b. Severe inflammation is seen in the joints of PBS-treated mice, as well as in the group of L-IVIg injected mice. Treatment with ACPA-sIVIg or H-IVIg prevented the development of substantial inflammation.
Figure 2.
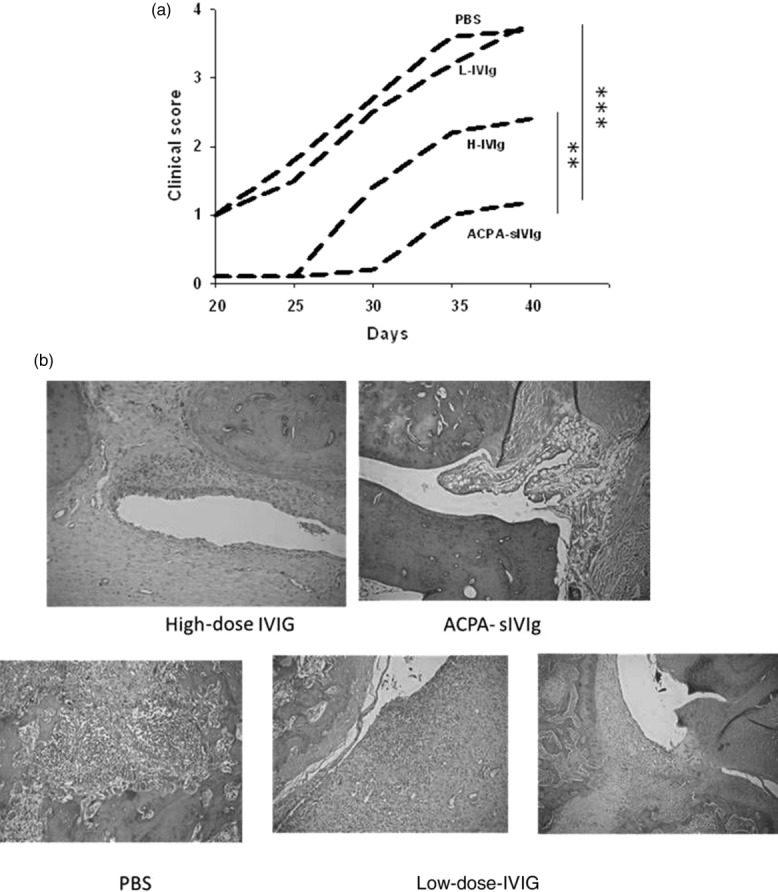
In-vivo analyses of the anti-citrullinated peptides antibody-specific intravenous immunoglobulin (ACPA-sIVIg)-treated mice. (a) Clinical score of collagen-induced arthritis (CIA) mice upon treatment with sIVIg, low- and high-dose IVIg and phosphate-buffered saline (PBS). The clinical score is described until day 40 after disease induction; n = 10 per group; **P < 0·03; ***P < 0·01. (b) Histological analysis of the joints. The joints were stained with haematoxylin and eosin (H&E). Severe inflammation is detected in PBS and low-dose IVIg-treated mice. See Supporting information, S3.
Suppression of the humoral response: lower sera titres of ACPA and anti-CII antibodies and the number of antibody-forming cells in the CIA mice treated with ACPA-sIVIg or H-IVIg
Circulating anti-type-II-collagen-(CII) antibody and anti-CCP titres were evaluated in the sera from CIA mice treated with ACPA-sIVIg, L-IVIg or H-IVIg and PBS. The titres of both mouse anti-collagen type II antibodies and ACPA were significantly lower in CIA mice treated with ACPA-sIVIg or high-dose IVIg compared to CIA mice that received low-dose IVIg or PBS (P < 0·002, P < 0·001, respectively) at day 38 (Fig. 3).
Figure 3.
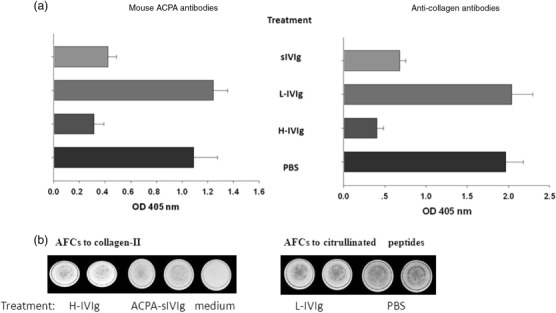
Humoral response in the collagen-induced arthritis (CIA)-treated mice. (a) Titres of anti-collagen-II antibodies and anti-anti-citrullinated peptides antibodies (ACPA) antibodies are presented upon different treatment with ACPA-specific intravenous immunoglobulin (sIVIg), high- and low-dose IVIg and phosphate-buffered saline (PBS). The results are expressed as optical density (OD) at 405 nm of three separate experiments. (b) Antibody-forming cell activity to collagen-II and citrullinated peptides are described at day 38 of the experiment when the mice were killed. The B-secreting antibody spots were counted. The numbers are presented with mean ± standard deviation (s.d.) in Table1.
We evaluated the activity of the antibody-forming cells (AFC) of the B cells isolated on magnetic beads from splenocytes of the ACPA-sIVIg, low- and high-dose IVIg and PBS-treated CIA mice at day 38 by ELISPOT assay. The data are presented as AFCs activity per 105 cells mean ± s.d.
We assessed whether the reduced sera titres of ACPA and collagen-II antibodies upon treatment with ACPA-sIVIg were related to the number of antibody-forming B cells or to lower potential to secrete antibodies. Data presented in Table1 show that ACPA-sIVIg treatment reduced the number of B cell-producing ACPA less than H-IVIg treatment (9 ± 2 AFC in comparison to 38 ± 3 AFC). L-IVIg treatment did not decrease the number of B cells AFC, 127 ± 22 AFC when compared to 147 ± 15 AFC of B cells purified from the PBS-treated groups of CIA mice (P > 0·05). Similar results were achieved when B cell AFC activity was tested with collagen-II. ACPA sIVIg reduced anti-collagen-II AFC activity when compared to PBS treatment or exposure to L-IVIg (P < 0·02). ACPA-sIVIG -AFCs was 4·2 times greater than H-IVIG, whereas no statistical significance was observed in the anti-collagen AFC activity in the ACPA-sIVIG-treated group of mice in comparison to the H-IVIG-treated group of mice. No statistical significance was noted in the anti-collagen-secreting cells between the AFC activity in the PBS-treated mice compared to AFC activity of the L-IVIg (P > 0·05).
Table 1.
Antibody-forming cell activity (AFC) upon treatment with anti-citrullinated peptides antibody-specific intravenous immunoglobulin (ACPA-sIVIg).
| Treatment | ACPA–AFCs 105 cells | Collagen-AFCs 105 cells |
| PBS | 147 ± 15 | 117 ± 7 |
| ACPA-sIVIg | 9 ± 2 | 26 ± 5 |
| P < 0.001 | P < 0.02 | |
| H-IVIg | 38 ± 3 | 49 ± 4 |
| P < 0.01 | P < 0.03 | |
| L-IVIg | 127 ± 22 | 108 ± 11 |
The data are presented as mean ± standard deviation of three separate experiments, as AFC per 105 counted cells. PBS = phosphate-buffered saline.
ACPA-sIVIg up-regulates expansion of T regulatory population in CIA mice
Recent reports have shown that IVIg treatment of CIA mice induced an increase in CD4+CD25+ FoxP3+ Tregs, as in other autoimmune diseases 12–15. Therefore, we examined the ability of ACPA-sIVIg treatment to cause an increase in the CD4+CD25+FoxP3+ Treg cell phenotype in CIA mice. The data presented in Fig. 4 show that ACPA-sIVIg enhanced the percentage of Tregs in CIA mice in the same manner as high-dose IVIg (P < 0·001) when compared with CIA mice treated with low-dose IVIg or PBS. No significant difference was noted in the percentage of Tregs in the splenocytes purified from high-dose IVIg and ACPA-sIVIg-treated mice (P > 0·05).
Figure 4.
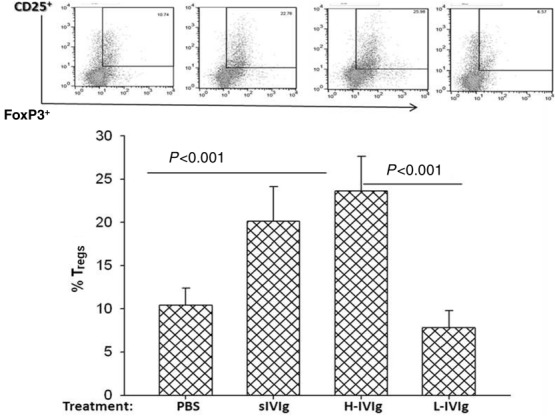
Regulatory T cells increase in the anti-citrullinated peptides antibody-specific intravenous immunoglobulin (ACPA-sIVIg)-treated collagen-induced arthritis (CIA) mice. The percentage of regulatory T cells (Tregs) CD4+CD25+forkhead box protein 3 (FoxP3) cells in the splenocytes of the mice treated with ACPA-sIVIg low- and high-dose IVIg and phosphate-buffered saline (PBS) was analysed by fluorescence activated cell sorter (FACS). The gating was on CD4+ T cells.
ACPA-sIVIg effect on the cytokine inflammatory network
We assessed the effect of ACPA-sIVIg treatment on the profile of proinflammatory and anti-inflammatory cytokines secreted by splenocytes in vitro. ACPA-sIVIg significantly inhibited the secretion of the proinflammatory cytokines TNF-α and IL-1β, as demonstrated in Fig. 5. The concentration of TNF-α in the culture fluid of splenocytes from ACPA-sIVIg-treated mice was 2·3 times less than secretion by splenocytes derived from PBS or low-dose IVIg-treated mice. No significant difference in TNF-α secretion was observed between the effect of ACPA-sIVIg treatment and high-dose IVIg treatments (P > 0·05). The concentrations of IL-1β in the culture fluid of splenocytes from ACPA-sIVIg and high-dose IVIg treatments were 3·6 and two times lower, respectively, in comparison to the IL-1β concentration in the culture fluid of the splenocytes from PBS or low-dose IVIg-treated CIA mice. ACPA-sIVIg enhanced secretion of anti-inflammatory cytokines by splenocytes in vitro. The concentrations of the anti-inflammatory cytokines IL-10 and TGF-β in the culture fluid of ACPA-sIVIg and high-dose IVIg-treated mice were increased significantly (P < 0·001 and P < 0·02, respectively) when compared with the IL-10 and TGF-β levels secreted by splenocytes originating from PBS and low-dose IVIg-treated mice.
Figure 5.
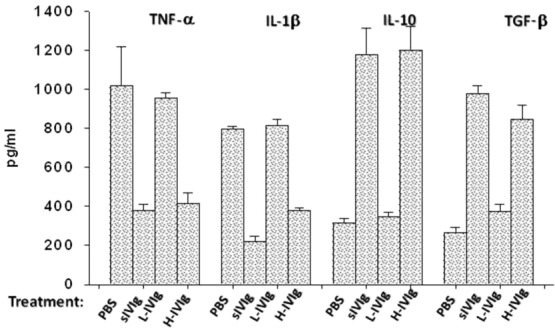
Cytokine profile expressed by the splenocytes in vitro. The levels of proinflammatory cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β and anti-inflammatory cytokines IL-10 and transforming growth factor (TGF)-β were studied by enzyme-linked immunosorbent assay (ELISA). The results are expressed as optical density (OD) at 405 nm of three separate experiments.
Discussion
RA is a systemic disease that leads to painful joint destruction and disability 1–5. The introduction of biological therapy, which targets specific elements of the immune system (e.g. anti-TNF, anti-IL-6 and kinase inhibitors), has revolutionized the treatment and outcomes of RA patients. Despite novel treatment strategies, not all patients respond well to therapy and a small number of RA patients remain partially or completely refractory to these and other conventional treatments. Biological treatments are cost-effective. For several decades, RA was considered to be a cell-mediated autoimmune disease based on the successful induction of the disease in severe combined immunodeficient (SCID) mice by T cells derived from patients with RA, and vaccination with T cells was proposed as a treatment 24–26. An additional revolutionary discovery was the diagnostic value of ACPA and their pathogenic role in RA 27–29. ACPA-positive RA patients suffer from a more severe disease, with more aggressive erosions and extra-articular manifestations, compared to ACPA-negative RA patients 30,31. APCA were found to be pathogenic and directed to synovial citrullinated proteins and play a significant role in the pathogenesis of RA. Therefore, these antibodies may be targets for therapy 1–5. Targeting the CD20 antigen by rituximab has been a successful therapeutic intervention in the treatment of RA 32–35. There is a basis to consider neutralization and immunomodulation of the pathogenic autoantibodies. Thus, IVIg for patients with RA has some advantages. IVIg has a wide range of immune modulatory activities with minimal side effects, and is no more expensive than most of the biological therapies. However, the mechanism by which IVIg immunomodulates the immune network in RA has not been completely clarified.
In our study, we have described one of the most important mechanisms for IVIg activity as an anti-idiotypic targeting and neutralizing agent. We show, for the first time, that the efficacy of anti-idiotypic anti-ACPA fraction of IVIg, named ACPA-sIVIg, inhibited ACPA activity in vitro. In addition, ACPA-sIVIg mitigated development of CIA in mice, which is a prototype model of RA and shares many clinical and histopathological similarities with RA patients 12,24. When injected into mice with CIA, ACPA-sIVIg led to a significant decline in the clinical joint inflammation score and was more effective than the entire IVIg preparation. Treatment with ACPA-sIVIg resulted in a more powerful decline in circulating anti-collagen and ACPA antibodies in comparison to the titres in mice treated with PBS or low-dose IVIg. Moreover, the decreased levels of autoantibodies correlated with less anti-collagen and ACPA antibody-forming cell (AFC) activity by the specific splenic B cells originating from CIA mice compared to AFC activity of B cells from the mice treated with PBS or low-dose IVIg. It was also evident that mice with CIA treated with ACPA-sIVIg demonstrated an increased percentage of Tregs compared to their percentage in the mice treated with total IVIg preparation. Furthermore, a favourable effect of ACPA-sIVIg on IVIg at the same concentration was shown by the elevated levels of anti-inflammatory cytokines (IL-10, TGF-β) and a decrease in the secretion of proinflammatory levels of cytokines (TNF-α, IL-1β) by splenocytes in vitro. Similar outcomes were evident in this study following treatment of mice with CIA with the high-dose IVIg regimen. However, this was not observed with a low-dose IVIg regimen or with PBS treatment. These results suggest that ACPA-specific IVIg may have a beneficial on the whole IVIg preparation for patients with RA, as well for those with other autoimmune disorders. Our results are in agreement with our previous studies treating autoimmune experimental models with anti-idiotypes directed to the pathogenic autoantibodies that improved disease scores. Interestingly, anti-idiotypic-sIVIg affected each autoimmune disease according to the animal presentation 17–21. In lupus mice, lupus-sIVIg inhibited the immune complex deposition in the mesangium, as well as reducing the glomerulonephritis. In APS mice, in addition to neutralizing the pathogenic antibodies, APS-sIVIg enhanced the expression of MMP2/MMP9, which enabled successful embryo implantation. In small-vessel vasculitis, myeloperoxidase-sIVIg reduced neutrophils’ oxidative burst, and in pemphigus mice pemphigus-sIVIg neutralized the anti-desmoglein-2/3 antibodies preventing the development of pemphigus.
We suggest that clinical cases that did not respond to IVIg may react to the disease-specific IVIg. This concept does not revoke other mechanisms of IVIg activities via Fab portion, such as expansion of Tregs, anti-cytokines, anti-chemokines, anti-receptors, adhesion molecules 14,36–39 or the role of sialylated Fc, which initiate an anti-inflammatory cascade through the lectin receptor-specific intercellular adhesion molecule-3 (ICAM-3) grabbing non-integrin-related-1 (SIGN-R1) or dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), which leads to up-regulated surface expression of the inhibitory FcR, FcgammaRIIb, on inflammatory cells, thereby attenuating autoantibody-initiated inflammation 40,41.
In summary, IVIg is an excellent preparation which, in the era of biological strategies, has additional wide-ranging capabilities, especially for those patients who do not respond to biological therapies.
Acknowledgments
We thank Dr Zera Telllier from Laboratoire du Fractionnement et des Biotechnologies (LFB), Les Ulis Cedex, France, for supporting this study. This work was performed in partial fulfilment of the requirements for a PhD degree by Nina Svetlicky, Sackler Faculty of Medicine, Tel Aviv University, and the Dr Pinchas Borenstein Talpiot Medical Leadership Program 2013, Sheba Medical Center, Tel-Hashomer, Israel.
Author contributions
All authors were involved in drafting the article and approved the final version to be published. Study conception and design: Y.S. and M.B. Experimental work was performed equally by N.S., S.K. and M.B; Q.O. helped in preparation of ACPA-sIVIg; S.G. performed the FACS analyses, H.A. collected the sera from RA patients; Y.S. participated in the sIVIG preparation, E.B.-M. and O.G. took part in performing the study. A.V. and I.B. are pathologists who covered the histological work.
Disclosures
The authors declare no conflicts of interest.
References
- Kuhn KA, Kulik L, Tomooka B, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–73. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal H, Bockermann R, Nandakumar KS, et al. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med. 2009;206:449–62. doi: 10.1084/jem.20081862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel C, Nogueira L, Laurent L, et al. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008;58:678–88. doi: 10.1002/art.23284. [DOI] [PubMed] [Google Scholar]
- Trouw LA, Haisma EM, Levarht EW, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009;60:1923–31. doi: 10.1002/art.24622. [DOI] [PubMed] [Google Scholar]
- Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014;16:R122. doi: 10.1186/ar4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrem A, Misra N, Hassan G, et al. Immunomodulation of autoimmune and inflammatory diseases with intravenous immunoglobulin. Clin Exp Med. 2005;5:135–40. doi: 10.1007/s10238-005-0079-y. [DOI] [PubMed] [Google Scholar]
- Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747–55. doi: 10.1056/NEJMra993360. [DOI] [PubMed] [Google Scholar]
- Kivity S, Katz U, Daniel N, Nussinovitch U, Papageorgiou N, Shoenfeld Y. Evidence for the use of intravenous immunoglobulins–a review of the literature. Clin Rev Allergy Immunol. 2010;38:201–69. doi: 10.1007/s12016-009-8155-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat C, Bertotto A, Ercolani R, et al. Long term treatment of rheumatoid arthritis with high doses of intravenous immunoglobulins: effects on disease activity and serum cytokines. Ann Rheum Dis. 1995;54:382–5. doi: 10.1136/ard.54.5.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumiati B, Casoli P, Veneziani M, Rinaldi G. High-dose immunoglobulin therapy as an immunomodulatory treatment of rheumatoid arthritis. Arthritis Rheumatol. 1992;35:1126–33. doi: 10.1002/art.1780351004. [DOI] [PubMed] [Google Scholar]
- Katz-Agranov N, Khattri S, Zandman-Goddard G. The role of intravenous immunoglobulins in the treatment of rheumatoid arthritis. Autoimmun Rev. 2015;14:651–8. doi: 10.1016/j.autrev.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Lee SY, Jung YO, Ryu JG, et al. Intravenous immunoglobulin attenuates experimental autoimmune arthritis by inducing reciprocal regulation of Th17 and Treg cells in an interleukin-10-dependent manner. Arthritis Rheumatol. 2014;66:1768–7. doi: 10.1002/art.38627. [DOI] [PubMed] [Google Scholar]
- Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. 2008;29:608–15. doi: 10.1016/j.it.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–89. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Luštrek M, Lorenz P, Kreutzer M, et al. Epitope predictions indicate the presence of two distinct types of epitope-antibody-reactivities determined by epitope profiling of intravenous immunoglobulins. PLoS One. 2013;8:e78605. doi: 10.1371/journal.pone.0078605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gunten S, Shoenfeld Y, Blank M, et al. IVIG pluripotency and the concept of Fc-sialylation: challenges to the scientist. Nat Rev Immunol. 2014;14:349. doi: 10.1038/nri3401-c1. [DOI] [PubMed] [Google Scholar]
- Shoenfeld Y, Rauova L, Gilburd B, et al. Efficacy of IVIG affinity-purified anti-double-stranded DNA anti-idiotypic antibodies in the treatment of an experimental murine model of systemic lupus erythematosus. Int Immunol. 2002;14:1303–11. doi: 10.1093/intimm/dxf099. [DOI] [PubMed] [Google Scholar]
- Blank M, Anafi L, Zandman-Goddard G, et al. The efficacy of specific IVIG anti-idiotypic antibodies in antiphospholipid syndrome (APS): trophoblast invasiveness and APS animal model. Int Immunol. 2007;19:857–65. doi: 10.1093/intimm/dxm052. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Feferman T, Meidler R, et al. A disease-specific fraction isolated from IVIG is essential for the immunosuppressive effect of IVIG in experimental autoimmune myasthenia gravis. J Neuroimmunol. 2008;194:89–96. doi: 10.1016/j.jneuroim.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Mimouni D, Blank M, Payne AS, et al. Efficacy of intravenous immunoglobulin (IVIG) affinity-purified anti-desmoglein anti-idiotypic antibodies in the treatment of an experimental model of pemphigus vulgaris. Clin Exp Immunol. 2010;162:543–9. doi: 10.1111/j.1365-2249.2010.04265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetlicky N, Ortega-Hernandez OD, Mouthon L, et al. The advantage of specific intravenous immunoglobulin (sIVIG) on regular IVIG: experience of the last decade. J Clin Immunol. 2013;33(Suppl. 1):S27–32. doi: 10.1007/s10875-012-9842-5. [DOI] [PubMed] [Google Scholar]
- Wegner N1, Lundberg K, Kinloch A, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233:34–54. doi: 10.1111/j.0105-2896.2009.00850.x. [DOI] [PubMed] [Google Scholar]
- Schellekens GA, de Jong BA, van den Hoogen FH, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson ME, Goldschmidt TJ, et al. Collagen induced arthritis as an experimental model for rheumatoid arthritis. Immunogenetics, pathogenesis and autoimmunity. APMIS. 1989;97:575–84. doi: 10.1111/j.1699-0463.1989.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Sakata A, Sakata K, Ping H, Ohmura T, Tsukano M, Kakimoto K. Successful induction of severe destructive arthritis by the transfer of in-vitro-activated synovial fluid T cells from patients with rheumatoid arthritis (RA) in severe combined immunodeficient (SCID) mice. Clin Exp Immunol. 1996;104:247–54. doi: 10.1046/j.1365-2249.1996.979670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eden W, Holoshitz J, Nevo Z, Frenkel A, Klajman A, Cohen IR. Arthritis induced by a T-lymphocyte clone that responds to Mycobacterium tuberculosis and to cartilage proteoglycans. Proc Natl Acad Sci USA. 1985;82:5117–20. doi: 10.1073/pnas.82.15.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Laar JM, Miltenburg AM, Breedveld FC, Cohen IR, de Vries RR. Towards T cell vaccination in rheumatoid arthritis. Chem Immunol. 1994;58:206–35. [PubMed] [Google Scholar]
- Agrawal S, Misra R, Aggarwal A. Autoantibodies in rheumatoid arthritis: association with severity of disease in established RA. Clin Rheumatol. 2007;26:201–4. doi: 10.1007/s10067-006-0275-5. [DOI] [PubMed] [Google Scholar]
- Vencovský J, Machácek S, Sedová L, et al. Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis. 2003;62:427–30. doi: 10.1136/ard.62.5.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Toes RE, Huizinga TW. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res Ther. 2005;7:R949–58. doi: 10.1186/ar1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesson C, Jacobsson LT, Sturfelt G, Matteson EL, Mathsson L, Rönnelid J. Rheumatoid factor and antibodies to cycliccitrullinated peptides are associated with severe extra-articular manifestations in rheumatoid arthritis. Ann Rheum Dis. 2007;66:59–64. doi: 10.1136/ard.2006.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani M, Anelli MG, Atzeni F, et al. Efficacy and safety of rituximab with and without methotrexate in the treatment of rheumatoid arthritis patients: results from the GISEA register. Joint Bone Spine. 2014;81:508–12. doi: 10.1016/j.jbspin.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Peterfy C, Emery P, Tak PP, et al. MRI assessment of suppression of structural damage in patients with rheumatoid arthritis receiving rituximab: results from the randomised, placebo-controlled, double-blind RA-SCORE study. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2014-206015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payet S, Soubrier M, Perrodeau E, et al. Efficacy and safety of rituximab in elderly patients with rheumatoid arthritis enrolled in a French Society of Rheumatology registry. Arthritis Care Res (Hoboken) 2014;66:1289–95. doi: 10.1002/acr.22314. [DOI] [PubMed] [Google Scholar]
- De Keyser F, Hoffman I, Durez P, Kaiser MJ2, Westhovens R2 MIRA Study group. Longterm followup of rituximab therapy in patients with rheumatoid arthritis: results from the Belgian MabThera in Rheumatoid Arthritis registry. J Rheumatol. 2014;41:1761–5. doi: 10.3899/jrheum.131279. [DOI] [PubMed] [Google Scholar]
- Kessel A, Ammuri H, Peri R, Blank M, Shoenfeld Y, Toubi E. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- Maddur MS, Othy S, Hegde P, et al. Immunomodulation by intravenous immunoglobulin: role of regulatory T cells. J Clin Immunol. 2010;30(Suppl. 1):S4–8. doi: 10.1007/s10875-010-9394-5. [DOI] [PubMed] [Google Scholar]
- Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2013;368:777. doi: 10.1056/NEJMc1215489. [DOI] [PubMed] [Google Scholar]
- Katz U, Shoenfeld Y, Zandman-Goddard G. Update on intravenous immunoglobulins (IVIg) mechanisms of action and off- label use in autoimmune diseases. Curr Pharm Des. 2011;17:3166–75. doi: 10.2174/138161211798157540. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–3. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol. 2010;30(Suppl. 1):S9–14. doi: 10.1007/s10875-010-9405-6. [DOI] [PubMed] [Google Scholar]