

Abstract
T helper type 17 (Th17) cells have been shown to be pathogenic in autoimmune diseases; however, their role in type 1 diabetes (T1D) remains inconclusive. We have found that Th17 differentiation of CD4+ T cells from BDC2·5 T cell receptor transgenic non-obese diabetic (NOD) mice can be driven by interleukin (IL)-23 + IL-6 to produce large amounts of IL-22, and these cells induce T1D in young NOD mice upon adoptive transfer. Conversely, polarizing these cells with transforming growth factor (TGF)-β + IL-6 led to non-diabetogenic regulatory Th17 (Treg17) cells that express high levels of aryl hydrocarbon receptor (AhR) and IL-10 but produced much reduced levels of IL-22. The diabetogenic potential of these Th17 subsets was assessed by adoptive transfer studies in young NOD mice and not NOD.severe combined immunodeficient (SCID) mice to prevent possible transdifferentiation of these cells in vivo. Based upon our results, we suggest that both pathogenic Th17 cells and non-pathogenic regulatory Treg17 cells can be generated from CD4+ T cells under appropriate polarization conditions. This may explain the contradictory role of Th17 cells in T1D. The IL-17 producing Treg17 cells offer a novel regulatory T cell population for the modulation of autoimmunity.
Keywords: AhR, IL-22, IL-23, TGF-β, Th17 subsets
Introduction
It has been shown that adoptive transfer of T helper type 17 (Th17) polarized BDC2·5 T cells leads to induction of type 1 diabetes (T1D) in non-obese diabetic severe combined immunodeficient (NOD.SCID) mice 1,2. However, in-vivo instability of these cells and their conversion to the Th1 phenotype in NOD.SCID mice precludes the conclusion that Th17 cells are involved directly in T1D pathogenesis 1–3. Thus, transfer of these cells to NOD mice 2 rather than NOD.SCID mice could possibly resolve the plasticity issue for further clarification of the role of Th17 cells in T1D. The adoptive transfer of Th17-polarized BDC2·5 cells that are stable in NOD mice induces pancreatic inflammation, but not T1D 2. Th17 cells are not a homogeneous population and different in-vitro conditioning could result in different subsets with a distinct cytokine profile. Investigation on the contribution of Th17 cells to pathogenesis in the disease model experimental autoimmune encephalomyelitis (EAE) has shown that Th17 cells derived by polarization with interleukin (IL)-23, IL-1β and IL-6 are pathogenic 4, while Th17 cells differentiated with replacement of IL-23 with transforming growth factor (TGF)-β are not able to induce disease 5. Differential expression of cytokines other than IL-17 or transcription factors in these subpopulations of Th17 cells might explain the disparity in pathogenic potential. Co-production of IL-10 and IL-17 might reduce the invasiveness of Th17 cells 5. We have shown previously that polarized Th17 cells from complete Freund's adjuvant (CFA) or bacillus Calmette–Guérin (BCG)-immunized NOD mice prevented adoptive transfer of disease 6.
IL-23 was shown to induce the expansion of a pathogenic Th17 cells from naive CD4 T cells in autoimmunity 7. However, other cytokines may be required for the optimal induction of these cells 4. As IL-6 induces IL-23R on T cells 8, we postulated that a combination of IL-23 and IL-6 may be able to provide alternative approach for the induction of pathogenic Th17 cells 9. In addition, TFG-β with IL-6 is normally able to induce Th17 cells 10. We therefore explored the induction of Th17 cells by IL-23 or TGF-β in the presence of IL-6 from naive CD4 T cells from T cell receptor transgenic BDC2·5 NOD mice. The BDC2·5 CD4 T cells are highly diabetogenic in NOD mice 11.
In this study, we generated two subpopulations of Th17 cells polarized by different conditions from BDC2·5 T cell receptor transgenic NOD mice. The Th17 cells induced by IL-23 + IL-6 cytokines were pathogenic upon adoptive transfer into young NOD mice. These pathogenic Th17 cells expressed the IL-22 gene differentially, and production of IL-22 in these cells was controlled by IL-23 in the polarizing cytokine combination. The non-pathogenic Th17 cells induced by TGF-β + IL-6 expressed differentially aryl hydrocarbon receptor (AhR) 12, IL-21 and IL-10 and much lower levels of IL-22. These cells did not induce diabetes upon adoptive transfer in NOD mice, but suppressed diabetogenic Th17 cells efficiently in vivo.
Materials and methods
Mice
NOD/Ltj and BDC2·5 T cell receptor (TCR) transgenic (Rag+/+ or Rag+/–) NOD mice used for these studies were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and were bred and housed in a pathogen-free environment at the animal care facility at Western University (London, Canada). All experiments were performed according to institutional guidelines and those of the Canadian Council for Animal Care. Mice were monitored for disease development by measuring urine glucose with Diastix strips (Bayer, Elkhart, IN, USA). Mice were considered diabetic after two consecutive positive (>11·5 mmol/l) urine glucose tests.
Cytokines and antibodies
Murine cytokines IL-6, IL-23 and TGF-β were purchased from BioLegend (San Diego, CA, USA). All cytokines were reconstituted and used according to the manufacturer's instructions. The following anti-mouse antibodies were purchased from BioLegend: anti-CD3ɛ (clone 145-2C11) was used to coat 24-well plates overnight in 1 ml sterile 1× phosphate-buffered saline (PBS) at 4°C; anti-CD28 (clone 37.51) was added to cultures on anti-CD3 coated plates; anti-interferon (IFN)-γ (clone XMG1.2) and anti-IL-4 (clone 11B11) were added to splenic or T cell cultures. Anti-TGF-β (clone ID11) was purchased from R&D Systems (Minneapolis, MN, USA).
In-vitro stimulation of splenocytes
Splenocytes from BDC2·5 mice were extracted and seeded into a 96-well plate at 2 × 105 cells per well with 1 μM PS3 mimotope peptide, SRLGLWVRME that stimulates BDC2·5 T cells 13. The PS3 peptide was synthesized, purified and characterized by mass spectrometry in our laboratory, as described previously 14. Cytokines were added at the following concentrations: IL-6 (20 ng/ml), IL-23 (20 ng/ml) and TGF-β (5 ng/ml), similar to the Th17 induction concentrations used by Sugita et al. 15. Where stated, the following antibodies were added to cultures: 5 μg/ml anti-IFN-γ, 5 μg/ml anti-IL-4 and 5 μg/ml anti-TGF-β. Cells were cultured for 4 or 5 days, as stated.
Naive T cell isolation
Splenocytes from BDC2·5 mice were extracted as above and naive T cells isolated using kits from Miltenyi Biotec (Auburn, CA, USA) to isolate CD4+CD62L+ cells according to the manufacturer's guidelines. Briefly, magnetic labelling of non-CD4+ T cells and separation using an LS column led to the depletion of non-CD4+ cells. Then positive selection of CD62L+ cells from this fraction was performed using an MS column to achieve a highly enriched (>90%) sample of CD4+CD62L+ cells. These cells were then washed, counted and plated at 3 × 106 cells per well in a 24-well plate that had been coated overnight with anti-CD3 and anti-CD28. Cells were cultured for 4 or 5 days, as stated, in complete RPMI [RPMI-1640 medium supplemented with 2 mM L-glutamine, 0.5% HEPES, 5 μg/ml penicillin, 100 U/ml streptomycin and 10% (v/v) fetal calf serum (HyClone Laboratories, Logan, UT, USA)].
For analysis of tissue lymphocytes by quantitative real-time reverse transcription–quantitative polymerase chain reaction (RT–qPCR), cells were extracted and restimulated as described above. Cells were plated at 3 × 106 cells per well on a 24-well plate that was coated overnight with 1 μg/ml anti-CD3 and cultured with 1 μg/ml anti-CD28 in 10% RPMI. After 48–72 h, supernatants were collected and RNA extracted for RT–qPCR analysis.
Enzyme-linked immunosorbent assay (ELISA)
Duoset ELISA kits from R&D Systems were used to analyse supernatants from in-vitro cultures for cytokines IL-10, IL-22, IL-17, IL-21 and IFN-γ. The manufacturer's protocols were followed directly. Standard curves were generated for each plate to determine sample concentration. Absorbance was determined using a Benchmark Microplate reader (BioRad, Hercules, CA, USA) and data were analysed using Microplate Manager version 4·0 software (BioRad). An ELISA kit from Biolegend was used for measurement of IL-9 concentration.
Proliferation assay
To determine cell proliferation, a tritiated thymidine uptake assay was performed. Splenocytes were plated in a U-bottomed 96-well plate at a density of 2 × 105 cells per well in culture medium containing various cytokines as stated. After 3 days of culture, 1 μCi of [3H]-thymidine was added to each well for 18 h. Cells were then harvested using a Tomtec cell harvester onto a Wallac filter (PerkinElmer, Waltham, MA, USA). Radioactivity was measured using a 1450 Microbeta liquid scintillation counter (PerkinElmer).
RNA extraction
For RNA extraction from splenocytes, lymph node cells or cultured lymphocytes, cells were disrupted in buffer RLT and β-mercaptoethanol and then homogenized by adding lysate to a QIAshredder spin column (Qiagen, Mississauga, ON, USA). Total RNA was then extracted using an RNeasy Mini Kit (Qiagen). Contaminating DNA was removed from all RNA samples using the DNase treatment and removal kit purchased from Ambion (Austin, TX, USA). The concentration of RNA in each sample was then determined by measuring absorbance at 260 nm using a NanoDrop 1000 spectrophotometer (NanoDrop Products, Wilmington, DE, USA).
qRT–PCR
For quantification of specific genes using qRT–PCR, 1–2 μg RNA from each sample was reverse-transcribed into first-strand cDNA using oligo dT12–18 primers from Superscript II Reverse Transcriptase Kit (Invitrogen, Carlsbad, CA, USA) and using a GeneAmp PCR System 2400 from Applied Biosystems (Foster City, CA, USA). Resultant cDNA was diluted in diethylpyrocarbonate (DEPC) water to a consistent concentration for each experiment, usually 225 ng/μl for RNA extracted from in-vitro cultures, and 500 ng/μl for whole tissues. cDNA was then amplified using Quantifast SYBR Green PCR Kit (Qiagen), according to the manufacturer's protocols. Gene-specific primers were purchased from Sigma-Aldrich (St Louis, MO, USA) and used at a concentration of 1·25 μM. Amplification was performed using a Corbett Rotor-Gene 6000 thermocycler (Corbett Life Sciences, San Francisco, CA, USA) and analysed using the manufacturer-provided Rotor-Gene software. The assay uses a two-step melting/annealing programme over 40 cycles of amplification. The Pfaffl method was used to quantify cycle threshold (Ct) data values, and all primers were determined to be 95–100% efficient.
Adoptive transfer
In the transgenic adoptive transfer model, splenocytes from BDC2·5 mice were cultured with the indicated cytokines and antibodies as well as PS3 mimotope as antigen for 4 days. Cells were then washed twice with sterile PBS to remove excess cytokines and resuspended in PBS. Five-week-old female NOD mice were then injected with 5 × 106 cells each in 200 μl intravenously (i.v.) through the tail vein. Mice were monitored for diabetes development every 3 days by urine glucose tests. The Kaplan–Meier survival estimate was used to determine differences in the treatment and control groups.
Statistical analyses
Statistical analysis was performed using GraphPad Prism 5·01 (La Jolla, CA, USA). Error bars represent standard error of the mean (s.e.m.) among samples. Significant differences between samples were determined using analysis of variance (anova) and Student's t-test, with P < 0·05 considered significant for all experiments; for these experiments, *P < 0·05, **P < 0·01 and ***P < 0·001.
Results
Polarization of naive BDC2·5 T cells into Th17 cell subsets
T cells can be induced to differentiate into a number of different effector subsets, each characterized by their cytokine production and other surface markers. In this study, naive CD4+CD62L+ T cells from spleens of BDC2·5 mice were isolated and stimulated with anti-CD3 and anti-CD28 antibodies as well as with polarizing cytokines and anti-IFN-γ and anti-IL-4 neutralizing antibodies to induce differentiation of Th17 cells. When naive BDC2·5 T cells are differentiated by the addition of TGF-β + IL-6 or IL-23 + IL-6, IL-17 production is significantly higher in Th17-polarized cells compared to non-polarized cells (Fig. 1, P < 0·001). The time–course of the secretion of IL-17 from days 1–5 suggests that the total amounts of IL-17 secreted by day 5 is similar under the two polarizing condition; however, IL-23 helps to secrete more IL-17 from the cells 16. With IL-23 + IL-6, a significant increase in the production of IL-22 is observed compared with TGF-β + IL-6 polarization (Fig. 1, P < 0·001). Such an increase is not seen, however, in the non-polarized cells.
Figure 1.
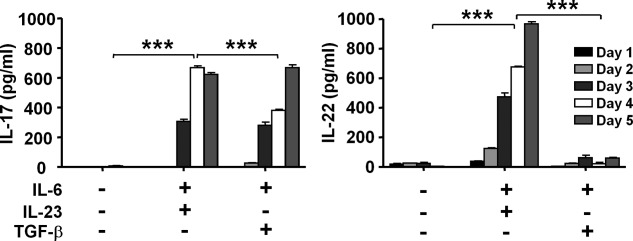
Polarization of naive BDC2·5 T cells into different T helper type 17 (Th17) subsets. Spleens and pancreatic lymph nodes were extracted from BDC2·5 mice and naive T cells were isolated by selecting for CD4+CD62L+ cells. These cells were then cultured in 24-well plates (3 × 106/well) that were coated with 5 μg/ml anti-CD3 and 5 μg/ml anti-CD28 antibodies overnight. Cells were polarized with 20 ng/ml interleukin (IL)−6, 5 ng/ml transforming growth factor (TGF)-β and/or 20 ng/ml IL-23 for 4 days. Anti-interferon (IFN)-γ (5 μg/ml) and anti-IL-4 (5 μg/ml) antibodies were added to all wells except the no cytokine control. Supernatants from cultures were collected and analysed for IL-22 and IL-17 expression from days 1–5 using enzyme-linked immunosorbent assay (ELISA). Results are shown as mean concentration ± standard error of the mean (s.e.m.). The analysis of variance (anova) test was used for the analysis and all values, with P < 0·05 considered significant (***P < 0·001).
Time–course of Th17 subset differentiation from BDC2·5 T cells
For characterization and optimization of Th17 cell differentiation, the Th17(IL-23 + IL-6) cell subset, Th17(TGF-β + IL-6) cell subset and a non-polarized subset were compared quantitatively in their ability to produce cytokines between 2 and 5 days after stimulation. Using qRT–PCR we found that both in Th17(IL-23 + IL-6) and Th17(TGF-β + IL-6) cells, the major Th17 cytokines IL-17 and IL-22 were produced significantly in the highest amounts on day 4 compared to all other days (Fig. 2, P < 0·001). IL-10 is produced in the highest amounts by Th17(TGF-β + IL-6) compared to non-polarized cells and Th17(IL-23 + IL-6) cells, which produced significantly lower amounts than all groups by day 5 (P < 0·001, P < 0·01, respectively) (Fig. 2). Similarly, IL-17 was shown to have the highest production on day 4 by cells polarized by TGF-β + IL-6, while the IL-23 + IL-6-induced cells can also produce copious amounts of IL-17 compared to the non-polarized group (P < 0·001). As expected, IL-22 production was considerably higher in IL-23 + IL-6 polarized cells than all other groups, and showed a greater than 1000-fold increase in IL-22 mRNA production on day 4 compared to the previous days (Fig. 2, P < 0·001). The IFN-γ production was measured to determine Th1-like cells. On day 4, IFN-γ production was significantly higher in the non-polarized group compared with both Th17-polarized cells. Interestingly, optimal IFN-γ production occurs by day 3 after stimulation, which suggests that cells can differentiate to a Th1 phenotype and produce effector cytokines more quickly than cells differentiating to a Th17 phenotype.
Figure 2.
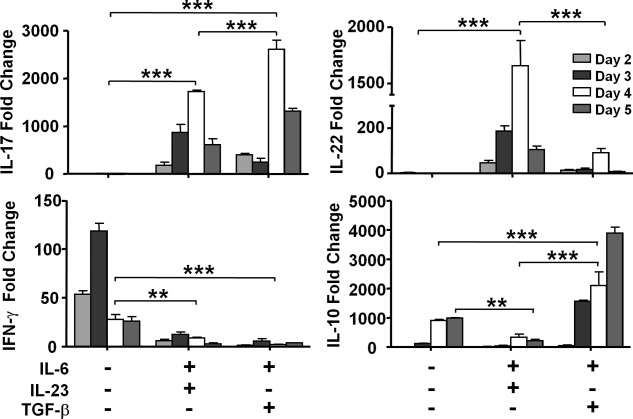
Differential expression of interleukin (IL)−22 and IL-10 by polarized T helper type 17 (Th17) subsets. Spleens and pancreatic lymph nodes were extracted from BDC2·5 mice and naive T cells were isolated by selecting for CD4+CD62L+ cells. These cells were then cultured in 24-well plates (3 × 106/well) that were coated with 10 μg/ml anti-CD3 and 2 μg/ml anti-CD28 antibodies overnight. Cells were polarized with 20 ng/ml IL-6, 5 ng/ml transforming growth factor (TGF)-β and/or 20 ng/ml IL-23 for 4 days. Anti-interferon (IFN)-γ (5 μg/ml) and anti-IL-4 (5 μg/ml) were also added to all wells except to the no cytokine control. Media and cytokines were refreshed after day 3. RNA was extracted from cells on each of days 2–5 and analysed by real-time reverse transcription–quantitative polymerase chain reaction (RT–qPCR). Relative results using beta actin as a housekeeping gene are shown as mean expression ± standard error of the mean (s.e.m.); all values with P < 0·05 were considered significant (**P < 0·01; ***P < 0·001).
IL-17 production shows somewhat inconsistent results using the qRT–PCR assay. Th17(TGF-β + IL-6) cells exhibit higher expression of IL-17 by qRT–PCR, whereas analysis of the supernatant points to significantly higher production of IL-17 by Th17(IL-23 + IL-6) cells (Fig. 1). This could be because IL-23 is known to help the secretion of IL-17 from the polarized Th17 cells 16. In addition, this may reflect the nature of the assays, as qRT–PCR analysis is fixed for equal amounts of cDNA, whereas the supernatant is affected by the number of cells. Therefore, Th17(IL-23 + IL-6) cells might demonstrate much higher proliferation than (TGF-β + IL-6) cells, as reflected by the assay. This was tested below in antigen-specific proliferation of the differentially polarized Th17 cells.
Antigen-specific proliferation of Th17 subsets derived from BDC2·5 T cells
As both TGF-β + IL-6 and IL-23 + IL-6 cytokine combinations were able to induce differentiation of BDC2·5 CD4 T cells to Th17 cells, we sought to determine if there was any difference between those two populations in terms of proliferation in response to antigen stimulation. The splenic BDC2·5 T cells were stimulated with mimotope peptide PS3 (1 μM) 14 for 5 days in the presence of TGF-β (3 ng/ml) + IL-6 (20 ng/ml) for Th17(TGF-β + IL-6) polarization, IL-23 (10 ng/ml) + IL-6 (20 ng/ml) for Th17(IL-23 + IL-6) polarization, or left non-polarized. Analysis of the proliferative capacity of these cells by [3H]-thymidine incorporation showed a significantly lower level of proliferation (Fig. 3) in the Th17(TGF-β + IL-6) [23 714 ± 554 counts per minute (cpm)] compared to that of Th17(IL-23 + IL-6) (65 013 ± 5916 cpm) or non-polarized cells (65 200 ± 6079 cpm). The background proliferation of cells with no antigen was < 2000 cpm in repeated experiments.
Figure 3.
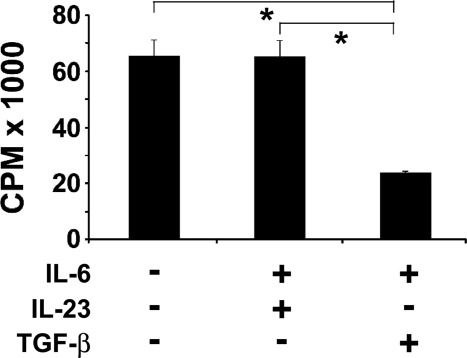
Differential proliferation of polarized T helper type 17 (Th17) subsets by antigen. Spleens were harvested from 6–8-week-old BDC2·5 non-obese diabetic (NOD) mice, and single-cell suspensions were prepared. The cells (2 × 105 cells/well) were cultured in 96-well plates in the presence of PS3 mimotope (1 µM) as antigen in complete RPMI-1640 medium at 37°C, 5% CO2 for 5 days. The T cells were either polarized with transforming growth factor (TGF)-β (3 ng/ml) + interleukin (IL)-6 (20 ng/ml) cytokines or IL-23 (10 ng/ml) + IL-6 (20 ng/ml) cytokine cocktails. Cells stimulated with PS3 mimotope without cytokine treatment were used as a positive control. After 72 h, [3H]-thymidine was added and incorporation of thymidine was measured and reported as mean counts per minute (cpm) ± standard error of the mean (s.e.m.) and all values with P < 0·05 considered significant (*P < 0·05). The background proliferation of cells with no PS3 mimotope as antigen was less than 2000 cpm in repeated experiments.
Cytokine, cytokine receptor and transcription factor gene expression in Th17 cells derived from BDC2·5 T cells
We analysed mRNA expression in Th17(TGF-β + IL-6) and Th17(IL-23 + IL-6) cells by real-time PCR to determine if specific cytokine, cytokine receptor and transcription factor gene expression exists in those two populations. Th17(IL-23 + IL-6) cells expressed significantly higher levels of IL-22 mRNA (Fig. 4). Accordingly, expression of the IL-23 receptor was significantly higher in Th17(IL-23 + IL-6) cells, indicating that these cells could be more responsive to IL-23 and hence produce more IL-22. When the cognate PS3 antigen is present the gene expression for IL-17 is similar under the two polarizing conditions, but expression of IL-22 was blocked in Th17(TGF-β + IL-6) cells by TGF-β. Differential mRNA expression was not limited to the IL-22 cytokine gene, as a number of other genes were expressed differentially between the two populations of BDC2·5 cells polarized with two cytokine cocktails (Fig. 4). The BDC2·5 cells polarized with TGF-β + IL-6 showed a significantly higher expression of aryl hydrocarbon receptor (Ahr) 12 mRNA compared to cells polarized with IL-23 + IL-6. IL-21 gene expression was restricted to Th17(TGF-β + IL-6) cells. Another significant difference in cytokine expression between Th17 populations was for IL-10 mRNA. While an 80-fold change in IL-10 mRNA expression has been observed in Th17(TGF-β + IL-6) cells compared to non-polarized cells, Th17(IL-23 + IL-6) cells showed a much smaller change in IL-10 mRNA level. A relatively higher level of IL-9 mRNA expression has been observed in Th17(TGF-β + IL-6) cells compared to Th17(IL-23 + IL-6). The highest level of IFN-γ mRNA was seen in non-polarized BDC2·5 cells stimulated with PS3 mimotope, and both Th17 populations showed a significantly lower level of IFN-γ gene expression. We also observed a significant increase in the expression of granulocyte–macrophage colony-stimulating factor (GM-CSF) mRNA in the pathogenic Th17(IL-23 + IL-6) cells.
Figure 4.
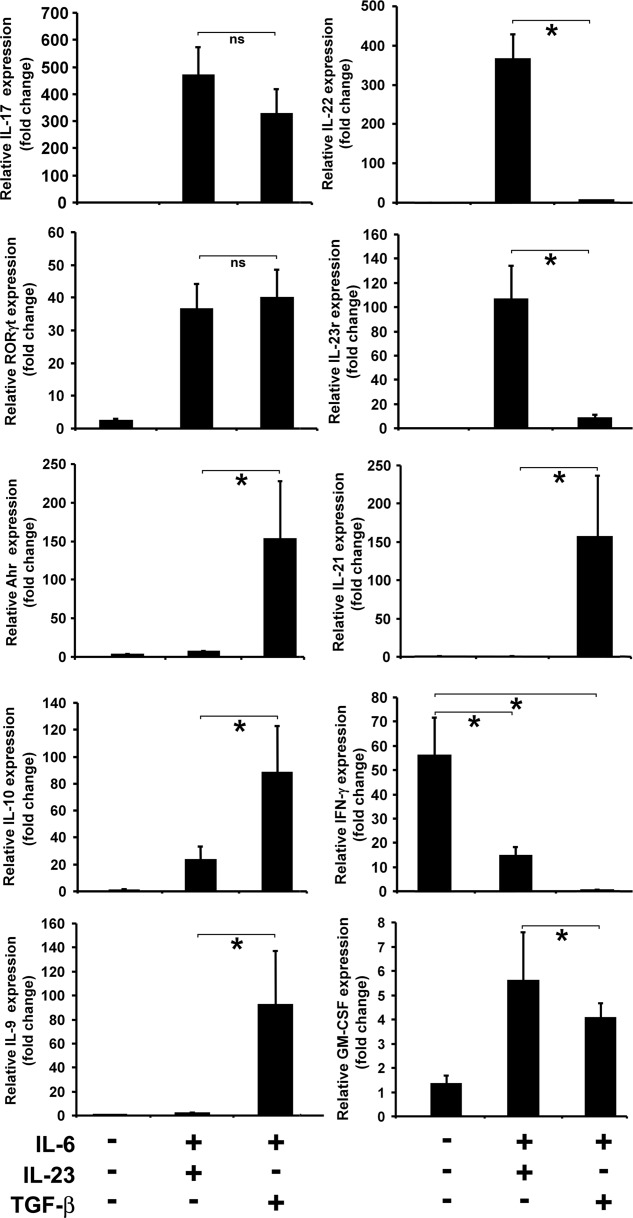
Polarization of BDC2·5 T cells with interleukin (IL)-23 + IL-6 or transforming growth factor (TGF)-β + 1L-6 induces T helper type 17 (Th17) subsets with differential cytokine, cytokine receptor and transcription factor profile. Differential gene expression was analysed in splenocytes from BDC2·5 non-obese diabetic (NOD) mice that were stimulated with PS3 mimotope and polarized with TGF-β + IL-6 or IL-23 + IL-6 for 5 days. Total RNA was extracted and transcribed into cDNA and real-time reverse transcription–quantitative polymerase chain reaction (RT–qPCR) performed using gene specific primers. The Pfaffl method was used for quantification of cycle threshold values using β-actin as a housekeeping gene. Results are shown as fold change in mRNA expression and are significantly different (*P < 0·05; ns = not significant), as indicated between various TGF-β + IL-6 and IL-23 + IL-6 groups, expect for granulocyte–macrophage colony-stimulating factor (GM-CSF) expression. Figure shows representative data from various reproducible experiments.
Functional response of Th17 subsets derived from BDC2·5 T cells after polarization with TGF-β + IL-6 or IL-23 + IL-6
To monitor the biological role of two populations of Th17 cells in vivo, BDC2·5 T cells from BDC2·5 NOD mice were stimulated with PS3 mimotope as antigen in the presence of either TGF-β + IL-6 or IL-23 + IL-6 cytokine cocktails for Th17 polarization. BDC2·5 splenocytes stimulated with PS3 mimotope 13 without cytokine polarization were used as control cells. After 5 days, cells were washed and injected into the tail vein of young 4–6-week-old NOD mice. Mice that received Th17(IL-23 + IL-6) cells began to develop diabetes within a week after adoptive transfer, and all of them were diabetic after 23 days (Fig. 5a) (P < 0·001). Adoptive transfer of Th17(TGF-β + IL-6) or activated non-polarized cells did not lead to diabetes in the recipient NOD mice in this time window.
Figure 5.
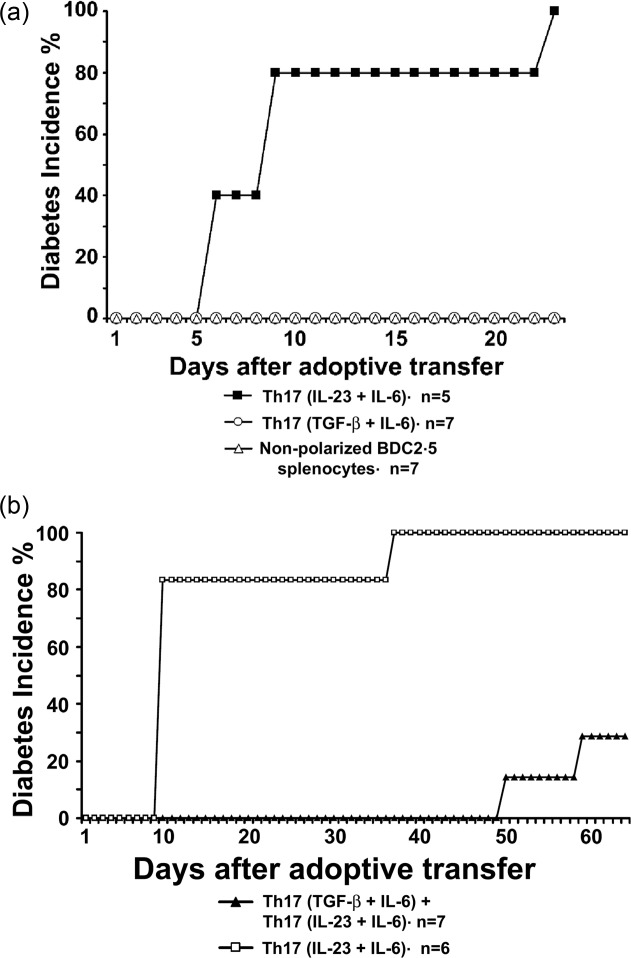
T helper type 17 (Th17) cells derived from BDC2·5 T cells by interleukin (IL)-23 + IL-6 polarization are highly diabetogenic, while regulatory T helper type 17 (Treg17) cells are induced by transforming growth factor (TGF)-β + IL-6. Spleens were harvested from 6–8-week-old BDC2·5 non-obese diabetic (NOD) mice, and single-cell suspensions were prepared. The cells (2 × 105 cells/well) were cultured in 96-well plates in the presence of PS3 mimotope (1 µM) in complete RPMI-1640 medium at 37°C, 5% CO2 for 5 days. Cells were either polarized with TGF-β (3 ng/ml) + IL-6 (20 ng/ml) cytokines or IL-23 (10 ng/ml) + IL-6 (20 ng/ml) cytokine cocktails. Cells stimulated with PS3 mimotope without cytokine treatment were used as a control. (a) Cells from various treatment groups were collected on day 5, washed and 5 × 106 cells/mouse were transferred adoptively via tail vein into 5-week-old NOD mice and the diabetes incidence was monitored. All mice injected with IL-23 + IL-6 polarized BDC2·5 cells developed diabetes. The Kaplan–Meier survival estimate was used to determine differences in the two groups. Significant difference was observed between the IL-23+IL-6 and TGF-β+IL-6 groups (P < 0·001). (b) Equal numbers (5 × 106) of Th17(TGF-β + IL-6) and Th17(IL-23 + IL-6) cells were injected into the tail vein of 5-week-old NOD mice. Th17(TGF-β + IL-6) cells blocked disease development. Significant difference was observed between the two groups (P < 0·01).
As Th17(TGF-β + IL-6) cells failed to induce disease, we wanted to determine if this population was able to affect the pathogenic ability of Th17(IL-23 + IL-6) cells in vivo. We transferred both populations together into young NOD mice. Mice transferred adoptively with Th17(IL-23 + IL-6) cells developed diabetes rapidly within a week, but co-transfer of Th17(TGF-β + IL-6) cells delayed (P < 0·01) disease development significantly (Fig. 5b). We observed that Th17(TGF-β + IL-6) cells were not only non-pathogenic in NOD mice, but played a regulatory role in the inhibition of pathogenic Th17 cells.
Discussion
We have found that two different protocols for Th17 cell differentiation give rise to two distinct cytokine profiles. The Th17 cells induced by TGF-β are functionally distinct from IL-23-induced Th17 cells. The Th17 cells polarized with IL-23 + IL-6 predominantly expressed IL-22 and were highly diabetogenic in the recipient NOD mice. The Th17 cells polarized with TGF-β + IL-6 expressed aryl hydrocarbon receptor, IL-10, IL-21 and IL-9 and were non-diabetogenic and were able to suppress pathogenic Th17 cells in co-adoptive experiments. Our results are supported by other studies, in that not all Th17 cells are pathogenic and Th17 cells generated with TGF-β and IL-6 cannot induce autoimmune disease without IL-23 17. Th17 cells are dependent upon IL-23 which, together with IL-6, produce pathogenic Th17 cells 7.
In our study the time–course of secretion of IL-17 and the expression of IL-17 gene from days 2 to 5 under the two polarizing conditions provided important new insight. The total amounts of IL-17 secreted by day 5 is similar under the two polarizing condition; however, IL-23 helps to secrete more IL-17 from the cells 16, despite the lower gene expression for IL-17. When the cognate PS3 antigen is present the gene expression for IL-17 is similar under the two polarizing conditions. The secretion and gene expression of IL-22 is completely dependent upon IL-23 and TGF-β suppresses the expression of IL-22. During the analysis of a variety of cytokine gene expressions in the Th17 polarized cells, we found that the IL-22 gene transcription and protein secretion were regulated positively in Th17(IL-23 + IL-6) cells as a function of the presence of IL-23 in the cytokine cocktail. While Th17(TGF-β + IL-6) cells highly expressed aryl hydrocarbon receptor (Ahr) 12, expression of this transcription factor in Th17(IL-23 + IL-6) cells is at the minimal level. Thus, TGF-β in the cytokine cocktail induces AhR expression in Th17 cells. IL-22 could be induced in the cells downstream of IL-23 receptor signalling and AhR activation. TGF-β down-regulates IL-22 production in Th17 cells through induction of the c-musculoaponeurotic fibrosarcoma (MAF) transcription factor 18. Increasing TGF-β concentrations in the cytokine cocktail reverses IL-22 production from Th17 cells polarized with IL-23 and IL-6. It has been shown that activation of AhR by an endogenous ligand or administration of its identified ligand, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), leads to the in-vivo expansion of regulatory T cells that suppresses EAE 19,20 and diabetes 21. Two mechanisms for this protective effect are proposed: the AhR agonist either expands regulatory T cells 19,20 or biases Th17 cells 22,23 towards a less invasive function. Thus, the increased expression of Ahr exclusively in Th17(TGF-β + IL-6) cells but not in Th17(IL-23 + IL-6) cells might explain their suppressive function, as Th17(TGF-β + IL-6) cells produce more IL-10 24.
The IL-10 gene expression is increased in Th17(TGF-β + IL-6) cells compared to Th17(IL-23 + IL-6) cells. It has been reported that TGF-β + IL-6 polarized Th17 cells showed a suppressive function and produced IL-10 when the concentration of TGF-β in the polarizing cytokine cocktail was increased 5. Moreover, we found that Th17 cells differentiated in the presence of TGF-β plus IL-6 express a high level of AhR 4, which promotes IL-10 production. These cells exhibited a regulatory function, as they produce IL-10 that is known to block the pathogenic Th17 cells and inhibit both differentiation and proliferation of Th17 cells 25. IL-10 also blocks GM-CSF production 26, and thus inhibits the functional role of pathogenic Th17 cells.
Compared to Th17(TGF-β + IL-6) cells, there was a higher level of IFN-γ production in Th17(IL-23 + IL-6) cells. However, IFN-γ gene expression was lowered drastically in both groups of IL-17-producing cells compared to non-polarized cells. The IFN-γ response could not be regarded as accountable for the pathogenic ability of Th17(IL-23 + IL-6) cells, as non-polarized cells with the highest IFN-γ response and Th1 phenotype were not able to induce diabetes in the recipient NOD mice within that time-frame.
The immunostimulation by a mycobacterial adjuvant that was shown to have a protective effect in NOD mice, while demonstrating an up-regulated response of IL-17, is further evidence that at least a subpopulation of Th17 cells could be regulatory 6. We have termed Th17(TGF-β + IL-6)-induced regulatory cells as Treg17 cells 27. Colonization of the gastrointestinal tract of NOD mice with the segmented filamentous bacteria (SFB) led to the increased level of IL-17 in the lamina propria and protection from diabetes 28. Recruitment of Th17 cells into gut lamina propria through CCR6–CCL20 interaction following strong immunostimulation was shown to play a critical role in acquiring a regulatory phenotype in Th17 cells 29. These studies have provided additional support to the conclusion that regulatory Th17 cells may exist in vivo and that there are distinct populations of Th17 cells in vivo beyond in-vitro polarizations 29. We have suggested previously the regulatory role of Th17 cells in autoimmunity 27,30. In Fig. 6 we propose a model in which IL-17 and IL-22 are ambivalent, and the presence of other cytokines or transcription factors modifies the overall function. The balance between two opposing cytokines, TGF-β and IL-23, determines the emergence of the regulatory or effector subsets of Th17 cells, respectively. TGF-β appears to promote both regulatory T cells and regulatory Th17 cells. Regulatory Th17 cells have a low proliferative capacity, low levels of IL-22 and higher levels of IL-10 and AhR that suppress pathogenic Th17 cells. Tipping the balance in favour of IL-23 by genetic or environmental cues leads to the more invasive and pathogenic Th17 cells that express higher levels of IL-22 and lower levels of AhR and IL-10 24. The contribution of each individual cytokine or transcription factor in driving Th17 cells to reveal pathogenic or suppressive potential remains to be determined. These studies indicate that acquiring regulatory function in Th17 cells is feasible and potentially effective for the immunoregulation of autoimmune diseases. Overall, our study shows that Th17 cells can have a pro- and anti-inflammatory role and Treg17 cells offer new ways to prevent and regulate autoimmunity. This is also supported by a recent study that indicated transdifferentiation of pathogenic Th17 cells into regulatory T cells in vivo, which is driven by TGF-β, and the resultant cells expressing high levels of AhR 31.
Figure 6.
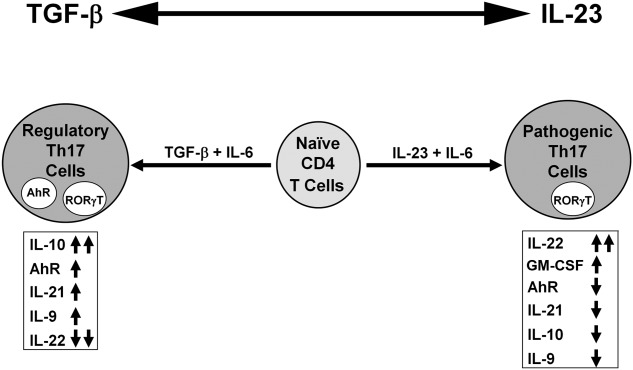
Proposed model for the induction of pathogenic and regulatory T helper type 17 (Th17) cell subsets. The overall balance between transforming growth factor (TGF)-β and IL-23 determines the emergence of the non-pathogenic regulatory (Treg17) and pathogenic Th17 cells. Elevated levels of interleukin (IL)-23 lead to the polarization of more invasive and pathogenic Th17 cells that express higher levels of IL-22. Increased amounts of TGF-β induced non-pathogenic Treg17 cells that express high levels of aryl hydrocarbon receptor (Ahr) and IL-10 and reduced level of IL-22.
Acknowledgments
Work in our laboratories was supported by grants from the Canadian Institutes of Health Research (CIHR).
Disclosure
The authors have no competing financial interests to declare.
References
- Bending D, De La PH, Veldhoen M, et al. Highly purified Th17 cells from BDC2.5 NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119:565–72. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–24. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Chung Y, Dong C. Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J. Immunol. 2009;182:2565–8. doi: 10.4049/jimmunol.0803931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–71. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)−17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Nikoopour E, Schwartz JA, Huszarik K, et al. Th17 polarized cells from nonobese diabetic mice following mycobacterial adjuvant immunotherapy delay type 1 diabetes. J Immunol. 2010;184:4779–88. doi: 10.4049/jimmunol.0902822. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima N, Mizoguchi I, Takeda K, Mizuguchi J, Yoshimoto T. TGF-beta is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23. Biochem Biophys Res Commun. 2009;386:105–10. doi: 10.1016/j.bbrc.2009.05.140. [DOI] [PubMed] [Google Scholar]
- Korn T, Mitsdoerffer M, Croxford AL, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:18460–5. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–7. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- Stockinger B, Di MP, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. 2014;32:403–32. doi: 10.1146/annurev-immunol-032713-120245. [DOI] [PubMed] [Google Scholar]
- Stadinski BD, Delong T, Reisdorph N, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–31. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoopour E, Sandrock C, Huszarik K, et al. Cutting edge: vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol. 2011;186:3831–5. doi: 10.4049/jimmunol.1003617. [DOI] [PubMed] [Google Scholar]
- Sugita S, Kawazoe Y, Imai A, Yamada Y, Horie S, Mochizuki M. Inhibition of Th17 differentiation by anti-TNF-alpha therapy in uveitis patients with Behcet's disease. Arthritis Res Ther. 2012;14:R99. doi: 10.1186/ar3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–9. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. 2013;252:116–32. doi: 10.1111/imr.12027. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107:20768–73. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet NI, Steppan LB, Vorachek W, et al. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–47. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di MP, Duarte JH, Ahlfors H, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity. 2014;40:989–1001. doi: 10.1016/j.immuni.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JH, Di MP, Hirota K, Ahlfors H, Stockinger B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLOS ONE. 2013;8:e79819. doi: 10.1371/journal.pone.0079819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoopour E, Bellemore SM, Singh B. IL-22, cell regeneration and autoimmunity. Cytokine. 2015;74:35–42. doi: 10.1016/j.cyto.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Huber S, Gagliani N, Esplugues E, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–65. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehler L, Foedinger M, Koeller M, et al. Interleukin-10 inhibits spontaneous colony-forming unit-granulocyte-macrophage growth from human peripheral blood mononuclear cells by suppression of endogenous granulocyte-macrophage colony-stimulating factor release. Blood. 1997;89:1147–53. [PubMed] [Google Scholar]
- Singh B, Schwartz JA, Sandrock C, Bellemore SM, Nikoopour E. Modulation of autoimmune diseases by interleukin (IL)-17 producing regulatory T helper (Th17) cells. Ind J Med Res. 2013;138:591–4. [PMC free article] [PubMed] [Google Scholar]
- Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108:11548–53. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues E, Huber S, Gagliani N, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–8. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoopour E, Schwartz JA, Singh B. Therapeutic benefits of regulating inflammation in autoimmunity. Inflamm Allergy Drug Targets. 2008;7:203–10. doi: 10.2174/187152808785748155. [DOI] [PubMed] [Google Scholar]
- Gagliani N, Vesely MC, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–5. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]