

Abstract
Delayed graft function (DGF) results from ischemia-reperfusion injury (IRI) and the generation of reactive oxygen species. We hypothesized that NADPH oxidase 2 (Nox2) plays an important role in pathways leading to DGF. We tested this hypothesis in vitro, in an animal model of IRI using wild type and Nox2−/− mice, and in patients with DGF. Under hypoxic conditions, primary tubular epithelial cells from Nox2−/− mice had reduced expression of MMP2, vimentin and HSP27. BUN and creatinine levels were significantly increased in both Nox2−/− and WT mice at 4 weeks and 6 months after IRI, suggesting the development of acute and chronic kidney injury. At 4 weeks, kidney fibrosis (α-SMA, picrosirius) and oxidative stress (dihydroethidine, HNE) were significantly reduced in Nox2−/− mice, confirming the oxidative and pro-fibrotic effects of Nox2. The molecular signature of IRI using genomic analyses demonstrated a significant decline in hypoxia reponse, oxidative stress, fibrosis, and inflammation in Nox2−/− mice. Immunohistochemical analyses of pre-implanatation kidney allograft biopsies from patients with subsequent DGF showed significantly greater Nox2 levels and vascular injury compared with patients without DGF. These studies demonstrate that Nox2 is a modulator of IRI and its absence is associated with reduced inflammation, OS, and fibrosis.
Introduction
Kidney transplantation is the ideal treatment modality for patients with end-stage-renal disease (ESRD) (1). In 2011, 10,589 kidney transplants were performed using deceased donor (DD) kidneys and 5,466 transplants with live donor kidneys (2). Graft outcomes for live donor kidney transplants are significantly better than DD kidney transplants because longer ischemic time results in greater ischemia and reperfusion injury (3). Ischemia-reperfusion injury (IRI) is universal in all DD kidney transplants and results from the imbalance between injury and repair. It has been associated with graft loss, delayed graft function (DGF) and rejection (4–7). DGF, which is defined as the need for dialysis within a week of transplantation, occurs in 20–40% of patients receiving DD kidneys and 5–10% of patient receiving live donor kidneys (4–7). DGF is the major factor affecting 1-year graft survival, and it is associated with tubulointerstitial fibrosis, morbidity, and mortality (6). Reactive oxygen species (ROS) and TGF-β1 have been implicated in the development of tubulointerstitial fibrosis following IRI (8, 9). ROS play a role in cellular defense, signaling, proliferation, differentiation, apoptosis and oxidative stress (OS) (10). Normally, a balance exists between ROS and antioxidant molecules that protect cells against oxidative damage. In the setting of IRI however, this delicate balance is disrupted and ROS damage carbohydrates, lipids, and nucleic acids, ultimately leading to cellular apoptosis and necrosis. Exogenous sources of ROS include toxins, drugs, and radiation while endogenous sources include the electron transport chain, peroxisomes, cytochrome P450 and the NADPH oxidases (10).
NADPH oxidases are ubiquitous transmembrane proteins that generate superoxide ions (O2−) from molecular Oxygen (O2) (11). Unlike the other ROS producers, the seven isoforms of NADPH oxidases produce superoxide ions as their primary product rather than a metabolic byproduct. NADPH oxidase 2 (Nox2) was first discovered in macrophages where it plays a role in generating the ROS used in the oxidative killing of bacteria and fungi. It was later discovered in other organ systems including the kidney (12). Deficiency of Nox2 is seen in individuals with chronic granulomatous disease and these patients suffer from recurrent infections mostly by catalase positive organisms including S. aureus and A. fumigatus (13).
We have previously demonstrated that Nox2 is involved in the pathogenesis of allograft fibrosis and chronic cyclosporine nephrotoxicity via inflammation and OS (14, 15). We have also shown that hypoxia induces Nox2 expression in NRK52E cells in the presence of TGF-β1. Based on these findings and the importance of Nox2 in phagocytes, we hypothesized that IRI is mediated in part by inflammation and OS driven by Nox2. We tested this hypothesis in vitro by investigating the effects of hypoxia on WT and Nox2−/− primary murine tubular epithelial cells (mPTECs). We also examined the effects of IRI in both WT and KO animals and finally, we evaluated the clinical association between pre-transplant Nox2 levels and the development of DGF.
Methods
Animal studies
Six to eight week old WT C57BL/6J (cat. # 664) and Nox2−/− B6.129S-CybbTm1Din/J (cat. # 2365) mice were purchased from Jackson Labs (Bar Harbor, Maine). All procedures were done in the surgical suites at the Wisconsin Institute of Medical Research in compliance with the institutional policies of the University of Wisconsin for the treatment and care of laboratory animals. WT and KO mice were randomized into 3 groups: Baseline, 4 week IRI, and 6 month IRI, with each group containing 5–8 animals. Animals in the IRI groups underwent a right nephrectomy under general anesthesia followed by clamping of the left renal artery for 30 minutes with a non-traumatic vascular clamp. At the end of the experiment, the clamp was removed and the wound was sutured after confirming reperfusion to the kidney. At the time of sacrifice, blood for serum analysis was collected by cardiac puncture and the remaining left kidney was harvested. BUN was measured on IdexxVetTest 8008 bioanalyzer (Idexx Laboratories, West Sacramento, CA) using compatible assay chips. Creatinine was measured at the NIH-sponsored O’Brien biochemical genetics laboratory at the University of Alabama using isotope dilution LCMS/MS.
Primary cell cultures
Four to seven day old WT and KO neonatal mice were euthanized in accordance with institutional guidelines for euthanasia of neonatal mice at the University of Wisconsin. Primary cell cultures from tubular epithelial cells were developed as previously described (16, 17). Briefly, kidneys were isolated and placed in conical tubes containing Dulbecco’s Modified Essential Media (DMEM; Gibco cat. # 11965-092) and 5% per volume fetal Calf Serum (FCS). The medulla was then carefully excised under a dissecting microscope and the cortex was placed in a pre-warmed (37°C), filter-sterilized, 1% solution of collagenase I (Worthington cat. # LS004196) and soybean trypsin inhibitor (Gibco cat. # 17075-029) in DMEM + 1% FCS. The cortices were minced finely then transferred to a 50 mL tube. Additional collagenase + trypsin inhibitor solution was added and the tubes were placed in a 37°C shaker at 225 rpm. After 20–30 minutes, the tubes were removed from the shaker then placed on ice for 2–3 minutes. The cell suspension was passed sequentially through a 250-micron mesh and an 80-micron mesh after which it was spun in a pre-cooled (4°C) centrifuge at 300×g for 5 minutes. The cell pellet was resuspended in pre-warmed (37°C), chemically defined renal epithelial growth medium (REGM; Lonza cat. # CC-3190). Finally, the cell suspension was placed in a collagen (Gibco cat. #A10483-01) coated 6-well plate (Thermo cat. # 140675) and placed in a humidified, 37°C, 21% O2, 5% CO2 incubator. Cells were allowed to grow undisturbed for 2 days before the media was changed. They were passaged on day 2 into two new collagen-coated plates (1:2) using 0.05% trypsin (Thermo, cat. # SH30236.01). On day 4, they were split into 4 new collagen-coated 6-well plates at a density of 350,000 cells per well. On day 5, they were approximately 70% confluent. Half of the plates were placed in a hypoxic incubator (37°C, 1% O2, 5% CO2) and the other half remained in the normoxic incubator (37°C, 21%O2, 5% CO2). On day 6 post-harvest, protein was extracted using 35 uL/well of 1ul/mL protease inhibitor cocktail (PIC; Sigma, cat. # P8340) in tissue protein extraction reagent (TPER; Thermo, cat. # 78510). The remainder of the extraction was performed as described previously (14, 18).
Patients
IRB approval for the use of patient data was obtained (UW Health Sciences IRB #2011-0127). A list of approximately 3000 kidney transplant patients who received their transplant between the years 2000 and 2010 was acquired from the University of Wisconsin transplant database. The patients were divided into groups who experienced delayed graft function (DGF) and controls. Patients who experienced a rejection episode within a year of transplantation were excluded. We identified 14 patients (6 with DGF and 8 controls) that matched these criteria who also had a pre-transplant donor kidney biopsy. Paraffin embedded biopsy samples were requested as per institutional guidelines and pathology Banff scoring was collected from the electronic medical record.
Antibodies and compounds
Nox2/gp91phox (1:250, BD Biosciences, San Diego, CA, 611414). α-SMA (1:2500 for WB, 1:50,000 for IHC, Sigma, St. Louis, MO, A2547). HNE (1:1000, Abcam, Cambridge, MA, ab46545). GAPDH (1:5000, Abcam, ab8245). MMP2 (1:500, Chemicon, Temecula, CA, AB809), Vimentin (1:500, Bioss, Woburn, MA, bs-0756R), HSP27 (1:500: #ab12351), Beta Actin (1:7500, US Biological, Salem, MA), Dihydroethidium (1:200, Life Technologies, Grand Island, NY, D23107), Phallotoxin (1 unit / 200 uL PBS, Life Technologies, A12379).
RNA extraction, purification and microarray analysis
RNA was extracted as previously described (14). 16 total mice were analyzed, 4 WT-baseline, 4 WT-IRI, 4 KO-baseline, 4 KO-IRI. RNA was analyzed on Affymetrix Mouse ST 2.0 microarray chips (Affymetrix, cat # 901169) using an Affymetrix GeneChip Analysis System. Mouse specific gene lists were generated from the rat genome database (rgd.mcw.edu). Gene groups included genes associated with oxidative stress, ischemia, hypoxia, inflammation, fibrosis (including interstitial fibrosis), matrix remodeling and reperfusion injury. Transcripts associated with signaling pathways were also included. Super-families under the headings of “hypoxia”, “fibrosis”, “oxidative stress” and “inflammation” were created to include related gene groups and signaling pathways. Analysis of the results was performed using Partek software.
Immunoblot analyses
Protein extraction was performed as previously described (14, 15, 19). Briefly, proteins were extracted from flash-frozen kidneys, and then 20ug of protein was run on an SDS-PAGE denaturing gel (BioRad cat #456-10260) at 200V. The blots were incubated with primary antibody overnight at 4°C and with secondary antibodies for 1 hour at RT. HRP was visualized using West Femto chemiluminescence kit (Pierce, cat# 34095) and images were acquired using a Fotodyne imaging system (Harlan, WI).
Immunohistochemistry
Immunohistochemical studies were performed as described previously (14, 15, 19). Briefly, kidney halves preserved in 10% formalin were paraffin embedded and cut into 5-µm thick sections. The slides were deparaffinized at 60°C for 30 minutes then rehydrated in xylene, a graded ethanol series to dH2O. For immunocytochemical studies, cells were grown on collagen coated circular coverslips in 24-well plates at a density of 80,000 cells per well. At the end of the experiment, cells were fixed with 4% paraformaldehyde for 10 minutes at RT. A 1:3 mixture of Prolong Gold antifade with and without DAPI (Life Technologies, cat. # P36931 and P36930 respectively) was used to mount the coverslips.
Picrosirius red staining was performed as described previously (20). For phalloidin staining, cell-coated coverslips were incubated with 0.1% TritonX in PBS for 5 minutes at −20°C. The coverslips were washed then incubated for 20 minutes at RT with 5µL methanolic solution of Alexa Fluor 594 Phalloidin (Life Technologies, cat. # A12381) in 20 µL PBS + 1% BSA per coverslip. For dihydroethidium staining, slides were incubated with 5 mM DMSO solution of dihydroethidium (Life Technologies, cat. # D23107) in PBS + 1%BSA (1:200) for 30 minutes at RT in the dark.
Digital Image Quantification
Slides were imaged using a Nikon Eclipse E600 equipped with an Olympus DP70 camera or a Leica DMIL microscope equipped with Leica EC3 camera. Depending on the size of the stained section, either the entire section or 5 random-field, non-overlapping images were taken for each section. Automated quantification was performed using a custom macro written for ImageJ software (NIH, imagej.nih.gov/ij/) as follows: the macro first opens the image then separates the image into 3 color components (using the Colour Deconvolution module from http://www.mecourse.com/landinig/software/cdeconv/cdeconv.html). The macro then selects the DAB component, performs thresholding, and then measures the thresholded area. If the image has significant empty space, an image component is thresholded to normalize for empty space. Before using the macro, the threshold level is optimized for the image set, and once determined, it is applied to the entire set of images in an experiment and there is no operator interference. The output is given as percent of total area, which is averaged then graphed in Prism.
Software
Data from microarrays were analyzed using Partek (Partek Inc., St. Louis, MO). Digital image quantification for Western blots and immunohistochemistry was done using ImageJ. Graphs and statistical analyses were done using GraphPad Prism (GraphPad Software, La Jolla, CA).
Statistics
Unpaired, 2-tail, 2-sample t-tests assuming equal variance were performed on semi-quantitative measurements of immunohistochemistry, and immunoblot images for each pair of comparison groups. Unpaired, 2-tail, 2-sample t-tests assuming equal variance was performed to compare renal function studies (BUN and creatinine) at an IRI time point (4 weeks or 6 months after) to baseline. ANOVA was used to determine a p-value for each gene in the microarray by comparing the up- and down-regulation of the biological replicates in the comparison groups. Genes were considered significantly different if the p value was < 0.05 and the fold difference was greater than 1.3. A Mann-Whitney test was used to determine the significance between WT and KO gene groups following IRI. Fischer’s exact test was used to determine the significance between cv scores in the DGF patients.
Results
Kidney function declined following IRI (Figure 1)
Figure 1.

IRI impaired renal function. Serum tests for renal function showed a steep decline in renal function (increased BUN and creatinine) 4 weeks after IRI. Renal function subsequently stabilized in the following months albeit at a significantly higher level than baseline. Nox2 knockout did not confer any improvement to renal function (WT baseline n=26, WT 4 weeks n=18, WT 6 months n=6, KO baseline n=25, KO 4 weeks n=17, KO 6 months n=6).
To determine the biological significance of our IRI model, we first examined kidney function in WT and Nox2−/− mice at baseline (WT, 26.2±0.9 mg/dL; KO, 20.4±0.5 mg/dL), 4 weeks (acute kidney injury), and 6 months after IRI (chronic kidney injury). Serum BUN significantly increased 4 weeks after IRI (WT 38.7±1.3 mg/dL, KO, 39.7±1.7 mg/dL; p<0.0001 for both) and stabilized at a higher level than baseline 6 months after injury (WT, 38.7±3.1 mg/dL, KO, 36.7±3.0 mg/dL; p<0.0001 for both). Similar results were observed for creatinine (Baseline: WT and KO, 0.11±0.01 mg/dL; 4 weeks after IRI: WT and KO, 0.19±0.01 mg/dL; p<0.0001 both; 6 months after IRI: WT 0.134±0.003, KO 0.132±0.007 mg/dL; p=0.001 for WT and p=0.012 for KO). Although lack of Nox2 was not associated with a significant change in BUN and creatinine levels, the observations indicated that IRI was associated with both acute and chronic decline in kidney function in this experimental model.
Absence of Nox2 was associated a decline in oxidative stress and fibrosis following IRI (Figures 2–4)
Figure 2.

Nox2 was induced by IRI. Semi-quantification of Nox2 immunostaining (DAB; Brown) showed increased staining 4 weeks after IRI compared to baseline. Nox2 levels dropped below baseline levels 6 months after IRI (n=4 for WT and KO at each time point). The Y axis in the right panel is a ratio (no units) of pixels. Specifically, it represents the number of pixels for Nox2 staining divided by number of pixels in the total image.
Figure 4.

(A) α-SMA was increased 4 weeks after IRI. Semi-quantification of immuonblots probing for α-SMA (42 KD) demonstrated higher protein levels of α-SMA 4 weeks after IRI compared to baseline. Nox2−/− significantly reduced protein levels of α-SMA. (B) Nox2 absence had no acute effect on lipid peroxidation. Semi-quantification of immunoblots probing for 4-HNE (70KD), a byproduct of lipid peroxidation, showed greater levels of 4-HNE 4 weeks and 6 months after IRI compared to Baseline. No difference was observed between KO and WT 4 weeks after IRI, while Nox2−/− significantly reduces 4-HNE levels 6 months after IRI in KO animals (n=3 for WT and KO at each time point, experiments repeated three times).
To determine whether Nox2 plays an important role in IRI-induced OS and fibrosis, we examined kidney sections from WT and KO animals stained with Nox2 (Figure 2), dihydroethidine and picrosirius red (Figure 3). We also completed immunoblot studies for α-SMA and 4-hydroxynonenal (4-HNE) (Figure 4). We noted a significant increase in Nox2 staining in WT animals at 4 weeks, with a decline to baseline levels by 6 months after IRI (Figure 2). As anticipated, there was no significant Nox2 staining in KO animals. We examined chronic renal injury using the Banff scoring model for tubular atrophy (ct), interstitial fibrosis (ci), and vasculopathy (cv) on H&E sections (21). These studies showed no statistical difference between the groups despite greater interstitial fibrosis in WT animals at 4 weeks (median ci score at 0.5 compared to 0, p=0.1). However, immunohistochemical analyses for dihydroethidine (OS) and picrosirius (fibrosis) demonstrated a similar pattern (Figure 3). Specifically, WT animals had a dramatic and significant increase in OS and fibrosis at 4 weeks, with a decline in staining by 6 months following injury. Immunoblot analyses confirmed these observations as α-SMA protein levels increased at 4 weeks and decline to near baseline by 6 months (Figure 4). Interestingly, these studies demonstrated significantly lower levels of α-SMA in Nox2−/− animals compared to WT at 4 weeks. 4-HNE, a marker of lipid peroxidation, was significantly increased 4 weeks after IRI compared to baseline in KO and WT animals (WT p=0.034; KO p=0.0015). There was no difference between WT and KO mice at that time point. However, lipid peroxidation continued to increase and was significantly higher 6 months after IRI compared to baseline (p=0.0076) and in WT compared to KO animals (p=0.013).
Figure 3.

Nuclear ROS and interstitial fibrosis are increased after IRI. Semi-quantification of fluorescence intensity showed higher nuclear ROS 4 weeks after IRI compared to baseline. Similar findings were seen with Pircosirius Red staining (inserts show higher magnification). Nox −/− was associated with significant reduction in nuclear ROS and Fibrosis. The kidneys remarkably healed 6 months after IRI (n=3 for WT and KO at each time point). The Y axis in panel B is a ratio (no units) of pixels. Specifically, it represents the number of pixels for DHE staining divided by number of pixels in the total image.
In aggregate, these studies suggest that Nox2 plays an important role in promoting OS and fibrosis in IRI, especially in the acute period.
Characterization of the molecular signatures for fibrosis, inflammation, OS, and hypoxia after IRI (Figure 5–6 and Tables 1–2)
Figure 5.

Gene groups of genes associated with hypoxia, oxidative stress, inflammation and fibrosis were increased by IRI. Heat maps of carefully curated gene groups were generated to show up- and down-regulation of transcripts associated with response to hypoxia, fibrosis and inflammation. Nox2−/− down-regulated transcripts associated with the same gene groups. Four animals in the WT-IRI group, 4 animals in the WT-t0 (baseline) group.
Figure 6.

IRI up-regulated genes associated with hypoxia response and inflammation. Only gene groups associated with response to hypoxia and inflammation were significantly up-regulated in WT animals relative to KO animals 4 weeks after IRI. Four animals in the WT-IRI group, 4 animals in the KO-IRI group.
Table 1. Molecular phenotype of fibrosis, OS, inflammation and hypoxia 4 weeks after IRI.
The molecular signature of IRI. 125 genes associated with fibrosis, oxidative stress, inflammation and hypoxia were mostly induced in WT mice 4 weeks after IRI.
| (a) Inflammatory response | (b) Hypoxia | (c) Oxidative Stress | (d) Fibrosis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N=56 | n=29 | N=16 | n=24 | ||||||
| Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change | Gene | Fold Change |
| Arrb2 | 1.45 | Irf1 | 1.37 | Aldoa | 1.30 | Akr1c18 | −3.04 | Cd44 | 2.40 |
| Axl | 1.45 | Itgb6 | −1.65 | Anxa1 | 1.61 | Aldh1a1 | 2.07 | Col1a1 | 1.40 |
| Bcl3 | 1.59 | Lck | 1.29 | Bhlhe40 | 1.49 | Apoa4 | −1.35 | Col1a2 | 1.50 |
| Birc3 | 1.48 | Lxn | 1.44 | Bhlhe41 | 1.73 | Apoe | 1.36 | Col4a3 | −1.43 |
| C3 | 2.79 | Ly86 | 2.07 | Birc2 | 1.48 | Arntl | −3.17 | Col6a6 | −1.55 |
| Ccl2 | 3.61 | Lyn | 1.34 | Cd14 | 2.00 | Car3 | −2.91 | Fn1 | 1.81 |
| Ccl3 | 1.99 | Map3k1 | 1.50 | Cd24a | 1.49 | Cygb | 2.07 | Itga4 | 1.37 |
| Ccl5 | 2.43 | Map3k14 | 1.31 | Cp | 2.06 | Hgf | 1.40 | Lamc2 | 1.36 |
| Ccl7 | 1.93 | Nfkb2 | 1.43 | Cpeb4 | −1.35 | Hspb1 | 1.79 | Mt1 | 1.35 |
| Ccl22 | 1.60 | Nfkbie | 1.46 | Csf1 | 1.34 | Ncf2 | 1.34 | Mt2 | 1.35 |
| Ccr1 | 1.34 | Nfkbiz | 1.39 | Dpp4 | −1.30 | Nqo1 | 1.58 | Mtss1 | 1.34 |
| Ccr2 | 2.50 | P2rx7 | 1.34 | Edn1 | 2.21 | Plk3 | −1.71 | Nphs2 | −1.45 |
| Ccr5 | 1.93 | Pik3cg | 1.56 | Fgg | 1.70 | Prnp | 1.41 | Pdgfra | 1.41 |
| Cd180 | 1.81 | Ptafr | 1.58 | Havcr1 | 7.11 | Rbm11 | 1.69 | Ren1 | −1.37 |
| Clec7a | 2.59 | Rps6ka4 | 1.42 | Hmox1 | 1.45 | Slc25a24 | 1.76 | Runx1 | 2.22 |
| Cfh | 1.55 | Sdc1 | 1.39 | Itgb2 | 1.63 | Txnip | 2.20 | Spp1 | 1.57 |
| Cxcl1 | 1.61 | Slc11a1 | 1.48 | Itpr2 | −1.32 | Tgfb1 | 1.33 | ||
| Cxcl2 | 1.48 | Syk | 1.31 | Kcnma1 | −1.34 | Thbs1 | 1.41 | ||
| Cxcl3 | 1.50 | Ticam2 | 1.35 | Lcn2 | 4.62 | Thbs2 | 1.92 | ||
| Cxcr6 | 1.78 | Tlr1 | 1.84 | Mmp14 | 1.85 | Tnc | 1.99 | ||
| Cxcl10 | 1.73 | Tlr2 | 1.34 | Mmp2 | 1.72 | Tnn | −1.33 | ||
| Cxcl11 | 1.36 | Tlr7 | 1.50 | Muc1 | 2.01 | Tnr | −1.41 | ||
| Ecm1 | −1.57 | Tlr8 | 2.32 | Myc | 1.66 | Trpv5 | 1.54 | ||
| Elf3 | 1.46 | Tlr9 | 1.51 | Ndrg1 | −1.44 | Vav3 | 1.31 | ||
| F2rl1 | 1.43 | Tlr11 | 1.31 | Nt5e | −1.36 | ||||
| Il1b | 1.42 | Tlr13 | 1.94 | Pld2 | 1.35 | ||||
| Il1f6 | 2.15 | Tnf | 1.31 | Pygm | −1.33 | ||||
| Il34 | 2.23 | Traf1 | 1.40 | Serpine1 | 1.87 | ||||
| Vcam1 | 3.80 | ||||||||
Table 2. Significant molecular changes in Nox2−/− vs. wild type mice after IRI.
Effects of Nox2 knockout on molecular signature of IRI. We identified 44 genes involved in fibrosis, oxidative stress, inflammation and response to hypoxia that appear to be associated with the Nox2 response to IRI. The majority of these genes were down-regulated.
| (a) Descriptive Table (alphabetical order) | |||||
|---|---|---|---|---|---|
| Gene symbol |
Fold change |
p | Gene symbol |
Fold change |
p |
| Inflammatory response (n=15) | Hypoxia (n=12) | ||||
| Axl | −1.38 | 0.021 | Bhlhe40 | −1.41 | 0.024 |
| Bcl3 | −1.40 | 0.026 | Cp | −1.84 | 0.044 |
| Ccl7 | −1.72 | 0.029 | Fgg | −1.68 | 0.012 |
| Ccr2 | −1.62 | 0.024 | Fos | −1.85 | 0.008 |
| Cfh | −1.54 | 0.035 | Itgb2 | −1.34 | 0.038 |
| Cxcl10 | −1.39 | 0.005 | Mmp14 | −1.85 | 0.014 |
| Cxcl3 | −1.43 | 0.031 | Ndrg1 | 1.51 | 0.014 |
| Ggt5 | −1.29 | 0.007 | Nt5e | 1.40 | 0.023 |
| Il34 | −1.30 | 0.032 | Plat | −1.39 | 0.041 |
| Itgb6 | 1.38 | 0.004 | Sox4 | −1.36 | 0.049 |
| Kit | −1.29 | 0.020 | Tfrc | 1.29 | 0.009 |
| Map3k1 | −1.31 | 0.043 | Vcam1 | −2.51 | 0.035 |
| Sdc1 | −1.30 | 0.047 | |||
| Tlr1 | −1.49 | 0.020 | |||
| Tlr6 | −1.52 | 0.024 | |||
| Fibrosis (n=11) | Oxidative Stress (n=6) | ||||
| Col1a1 | −1.79 | 0.026 | Akr1c18 | 2.18 | 0.009 |
| Col1a2 | −1.58 | 0.047 | Aldh1a1 | −1.48 | 0.024 |
| Col5a1 | −1.32 | 0.013 | Apoe | −1.66 | 0.012 |
| Ctgf | −1.31 | 0.038 | Car3 | 2.22 | 0.011 |
| Dcn | −1.43 | 0.046 | Cygb | −1.72 | 0.037 |
| Fn1 | −1.94 | 0.021 | Hspb1 | −1.54 | 0.039 |
| Itga4 | −1.35 | 0.035 | |||
| Mas1 | 1.49 | 0.002 | |||
| Spp1 | −1.53 | 0.041 | |||
| Trpv5 | −1.36 | 0.024 | |||
| Vav3 | −1.35 | 0.027 | |||
| (b) Upregulation and downregulation of genes after IRI in Nox2−/− compared to wild type mice |
|---|
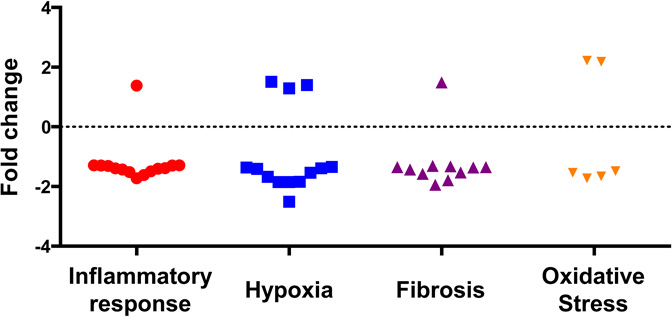 |
To investigate the effects of IRI and Nox2 in the acute phase, we determined gene expression profiles of fibrosis, inflammation, OS and hypoxia using microarray analyses in WT and Nox2−/− kidney tissues at baseline and 4 weeks after IRI. Genes was considered to be differentially expressed if the p value was < 0.05 and the fold difference was greater than 1.3.
A total of 125 genes involved in fibrosis (n=24), OS (n=16), inflammation (n=56) and hypoxia (n=29) were up- or downregulated in WT animals 4 weeks after IRI [WT-IRI] compared to baseline [WT-t0] (Figure 5, Table 1 and supplemental Table S1). Reproducibility of gene expression changes can be analyzed in the heat maps. Out of the 8 animals used in this analysis, one appears to be an outlier for all four-gene groups, the top animals (pink box) in the IRI group. For these animals, genes that increased expression (red) for the other three decreased expression or stayed the same for this animal (represented as blue or grey areas). Figure 6 compares within-group changes in genes involved in inflammation, hypoxia, fibrosis and OS between WT and KO animals. The analyses indicate that Nox2 primarily enhances inflammation and hypoxia responses after IRI.
Importantly, 44 genes (inflammation=15, hypoxia=12, fibrosis=11, and OS=6) were significantly up- or downregulated in Nox2−/− animals compared to WT after IRI (Table 2). Specifically, inflammatory genes Tlr1, Il34, Cxcl3, Ccr2, Ccl7, Cfh and Map3k1 involved in signaling and recruitment of inflammatory cells were reduced. Similarly, fibrosis genes including collagen, CTGF and fibronectin, hypoxic injury response and angiogenesis genes including fos, sox4, mmp14, plat, vcam1, and OS genes including axl, cygb and hspb1 (HSP27) were also significantly reduced in KO animals. Panel b demonstrates that most genes (n=37) were downregulated while a few (n=7) were upregulated in Nox2−/− animals.
In summary, the genomic studies suggest that Nox2 is involved in the pathogenesis of OS, hypoxia, inflammation, and fibrosis in the acute phase after IRI.
Absence of Nox2 was associated with reduced Vimentin, MMP2, and HSP27 expression during hypoxia in mouse primary tubular epithelial cells (mPTEC; Figures 7–8)
Figure 7.

Phalloidin staining was increased following hypoxia. Phalloidin or phallotoxin specifically stains F-Actin, which is a marker for matrix deposition. Phalloidin staining was significantly increased in both WT and Nox2−/− cells in hypoxic conditions.
Figure 8.

The absence of Nox2 was associated with a decrease in fibrosis and oxidative stess-associated protein expression in vitro during hypoxia. MMP, Vimentin, and total HSP27 were significantly decreased in KO cells following hypoxia.
Nox2 is primarily expressed by phagocytes but can also be up-regulated in renal tubular cells under stress (10, 14, 15). To determine the significance of Nox2 in tubular epithelial cell injury in hypoxia, mPTEC cells were incubated in 1% and 21% oxygen chambers. We first evaluated F-actin expression using phalloidin staining and determined that hypoxia was associated with a significant increase in stress fibers (WT, p=0.0051 and KO, p=0.0049), regardless of the presence of Nox2 (Figure 7). In contrast, vimentin, MMP2 and HSP27 were all significantly attenuated in KO cells following hypoxia, suggesting that despite the lack of effect on actin stress fibers, Nox2 regulates hypoxia-induced matrix remodeling in tubular epithelial cells.
Nox2 was significantly increased in DGF, and correlated with higher chronic vascular injury (Figure 9)
Figure 9.

Pre-transplant Nox2 and cv score predict DGF status. Nox2 was significantly correlated with DGF status compared to controls. Cv score in the same group of patients showed that a cv score ≥1 correlates with DGF status.
To further assess the clinical relevance of our studies and investigate the clinicopathlogical correlation between Nox2 and DGF, we compared Nox2 expression in pre-transplant biopsies of allograft recipients who later experienced DGF (n=6) to those who did not (n=8). Gender (2 vs. 4 female), and mean age (46.1±13 vs. 46.9±9 years) were not significantly different between the two groups. However, as expected per design, donor type (6 donor after cardiac death in the DGF groups vs. none in the control group) and cold ischemia times (16.6±5.7 vs. none in live donors in the control group) were different between the two groups.
Immunohistochemical analyses demonstrated that Nox2 was significantly higher (p=0.032) in the DGF group compared to controls. Furthermore, patients who experienced DGF were more likely to have cv Banff scores of 1 or more compared to controls (p=0.026), suggesting that Nox2 plays a role in the pathogenesis of IRI and DGF after kidney transplantation.
Discussion
Findings from this study demonstrate that Nox2 is involved in the pathogenesis of ischemia reperfusion injury. Specifically, our observations from in vitro, animal and clinical studies suggest that Nox2 absence prevents inflammation, OS and fibrogenesis following IRI and imply that Nox2 may be a valid therapeutic target for prevention of DGF and IRI-induced fibrosis.
Despite the decline in the rate of acute rejection, the incidence of DGF has remained nearly unchanged at approximately 25% in deceased donor kidney transplants (21). Challenges to understand the cause of DGF depend on donor and recipient variables and include IRI, the innate immune response and the adaptive immune response (22). An under-explored area that may hold great potential to improve transplantation outcomes is the design of novel strategies to apply specifically to organs to reduce intra-graft inflammation (23). Controlling the activation of innate immunity/inflammatory responses has been shown to be a promising strategy to increase the graft acceptance and survival (23). Nox2 inhibition strategies may offer an opportunity in this field (24).
Nox2 is the classical phagocytic Nox and its primary role is the generation of free radicals. Given the importance of phagocytes in IRI, we hypothesized that Nox2 inhibition could prevent kidney injury and DGF after ischemia-reperfusion. Although we did not observe a significant tubular expression of Nox2 in these studies, our findings are consistent with previous reports demonstrating that Nox2 inhibition attenuates cyclosporine-induced nephrotoxicity and allograft fibrosis, two kidney injury models in which inflammation plays a key role in fibrogenesis (14, 15). Our observations are also in agreement with studies demonstrating the association of inflammation and leukocyte recruitment with tissue fibrosis (25–27). However, our findings are in disagreement with a recent report demonstrating no significant role for Nox2 in obstructive nephropathy (28). The discrepancy might be explained by the difference in models of acute injury, although the unilateral ureteral obstruction model is also associated with significant involvement of phagocytes. It might also be explained by the limited biomarkers of fibrosis (picrosirius) and OS (8-oxo-DG) utilized in the study.
Genomic studies suggested that the antifibrotic effect of Nox2 inhibition was primarily mediated through inflammation and hypoxia genes. Consistent with this observation, there were more inflammation (n=14) and hypoxia-response (n=9) genes downregulated compared to OS (n=5 genes). Similarly, within-group changes for genes involved in inflammation and hypoxia were significantly narrower in Nox2−/− animals, indicating that the absence of Nox2 reduces the amplitude of inflammatory and hypoxia responses. Because only a limited number of genes are involved in the pathways affected by Nox2, the differences in gene expression between the WT and KO during IRI were not strong, so a low cutoff for fold change was used to define differential expression between these groups. Nevertheless, Nox2 remains an important source of ROS in the kidney (14, 15, 29, 30) and it is highly possible that it contributes to IRI via OS. It should also be noted that OS, hypoxia, and inflammation are closely related profibrotic processes and many genes arbitrarily classified in one category are in fact involved in several pathways.
Originally named gp91phox, Nox2 is the classical phagocytic NADPH oxidase (11, 12), an enzyme that is naturally involved in the immune response including the “oxidative burst” (11, 12). Nox2 is also the original identified example of a system that generates ROS as the primary function of the enzyme and not as a byproduct like the mitochondrion or peroxisomes. It is one of the seven currently known Nox isoforms, is constitutively associated with the transmembrane stabilizing protein p22phox and requires the recruitment of cytosolic components p47phox, p67phox, and p40phox for function (11). We have selected this isoform over the other isoforms for three reasons. First, apocynin and DPI, the most commonly used inhibitors of Nox are not specific to Nox or its isoforms and have global antioxidant characteristics (31, 32). The availability of Nox2 null mice (compared to other isoforms) will enable us to rigorously test our hypotheses in vivo. Furthermore, the in vivo use of Nox2 siRNA is limited by the off target effects and short half-life of the molecule. Second, we have been unable to detect measurable levels of other Nox proteins in renal tubular epithelial cells undergoing fibrogenesis in vitro, while we have repeatedly demonstrated that Nox2 protein is induced in tubular cells under similar conditions (14, 15, 33). Third, the functions of these isoforms may be different and cell/tissue specific (34–36) and our earlier data suggest that Nox2 plays a greater role in kidney allograft fibrosis and CNI-induced renal hypoxia and fibrogenesis (14, 15, 33).
We observed a near resolution of kidney injury despite reduced renal function by six months post IRI. This is in agreement with reports demonstrating accelerated healing in mice subjected to unilateral nephrectomy followed by IRI (37), and consistent with the strength of repair mechanisms in young and otherwise healthy mice. Repair mechanisms are not as potent in older kidneys and patients with multiple comorbidities. Notably, DGF is associated with significant medical and surgical complications in clinical transplantation, further outlining the importance of preventive and therapeutic options in this area. In conclusion, our findings demonstrate that Nox2 is a modulator of IRI and its absence is associated with reduced inflammation, OS, and fibrosis. These observations suggest that specific Nox2 strategies could be considered for the prevention and/or treatment of IRI and DGF.
Supplementary Material
Acknowledgements
Funding for this project was provided by the NIDDK grant (5R01DK092454-04) as well as generous funding from the University of Wisconsin department of medicine. Funding for the year of research for Aos Karim was through Howard Hughes Medical Fellows program.
Abbreviations
- 4-HNE
4 hydroxynonenal
- α-SMA
Alpha smooth muscle actin
- BSA
Bovine serum albumin
- BUN
Blood urea nitrogen
- Cv score
Chronic vascular injury score
- DAB
3,3'-Diaminobenzidine
- DAPI
4',6-diamidino-2-phenylindole
- DD
Deceased donor
- DGF
Delayed graft function
- DHE
Dihydroethidium
- DMEM
Dulbecco’s modified essential medium
- DPBS
Dulbecco’s phosphate buffered saline
- ESRD
End stage renal disease
- FCS
Fetal calf serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HRP
Horseradish peroxidase
- IHC
Immunohistochemistry
- IRI
Ischemia-reperfusion injury
- LCMS
Liquid chromatography mass spectroscopy
- mPTECs
Murine Primary tubular epithelial cells
- MMP2
Matrix metalloproteinase 2
- MS
Mass spectroscopy
- NADPH
Reduced Nicotinomide adenosine diphosphate
- Nox
NADPH oxidase
- NRK52E
Normal rat kidney 52E
- OS
Oxidative Stress
- REGM
Renal epithelial growth medium
- ROS
Reactive Oxygen Species
- RT
Room Temperature
- SDS-PAGE
Sodium dodecyl sulfate – polyacrylamide gel electrophoresis
- TBS
Tris(hydroxymethyl)amino-methane Buffered Saline
- TNF
Tumor necrosis factor
- TPER
Tissue protein extraction
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional Supporting Information may be found in the online version of this article, including: • Supplemental Table S1 (Gene names and fold change after IRI in WT mice)
References
- 1.Abecassis M, Bartlett ST, Collins AJ, Davis CL, Delmonico FL, Friedewald JJ, et al. Kidney transplantation as primary therapy for end-stage renal disease: A National Kidney Foundation/Kidney Disease Outcomes Quality Initiative (NKF/KDOQI (TM)) conference. Clin J Am Soc Nephro. 2008;3(2):471–480. doi: 10.2215/CJN.05021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Organ Procurement and Transplantation Network. OPTN/SRTR 2011 Annual Data Report: Kidney.3 [Google Scholar]
- 3.Kayler LK, Magliocca J, Zendejas I, Srinivas TR, Schold JD. Impact of cold ischemia time on graft survival among ECD transplant recipients: a paired kidney analysis. Am J Transplant. 2011;11(12):2647–2656. doi: 10.1111/j.1600-6143.2011.03741.x. [DOI] [PubMed] [Google Scholar]
- 4.Abreu SC, Goldfarb DA, Derweesh I, Thornton J, Urbain JL, Mascha E, et al. Factors related to delayed graft function after laparoscopic live donor nephrectomy. J Urology. 2004;171(1):52–57. doi: 10.1097/01.ju.0000098819.79442.ce. [DOI] [PubMed] [Google Scholar]
- 5.Leggat JE, Jr, Ojo AO, Leichtman AB, Port FK, Wolfe RA, Turenne MN, et al. Long-term renal allograft survival: prognostic implication of the timing of acute rejection episodes. Transplantation. 1997;63(9):1268–1272. doi: 10.1097/00007890-199705150-00013. [DOI] [PubMed] [Google Scholar]
- 6.Quiroga I, McShane P, Koo DDH, Gray D, Friend PJ, Fuggle S, et al. Major effects of delayed graft function and cold ischaemia time on renal allograft survival. Nephrol Dial Transpl. 2006;21(6):1689–1696. doi: 10.1093/ndt/gfl042. [DOI] [PubMed] [Google Scholar]
- 7.Troppmann C, Gillingham KJ, Benedetti E, Almond PS, Gruessner RWG, Najarian JS, et al. Delayed Graft Function, Acute Rejection, and Outcome after Cadaver Renal-Transplantation - a Multivariate-Analysis. Transplantation. 1995;59(7):962–968. doi: 10.1097/00007890-199504150-00007. [DOI] [PubMed] [Google Scholar]
- 8.Chen SW, Kim M, Song JH, Park SW, Wells D, Brown K, et al. Mice that overexpress human heat shock protein 27 have increased renal injury following ischemia reperfusion. Kidney Int. 2009;75(5):499–510. doi: 10.1038/ki.2008.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. American journal of physiology Renal physiology. 2011;301(2):F436–F442. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djamali A. Oxidative Stress as a Common Pathway to Chronic Tubulointerstitial Injury in Kidney Allografts. Am J Physiol Renal Physiol. 2007;293(2):F445–F455. doi: 10.1152/ajprenal.00037.2007. [DOI] [PubMed] [Google Scholar]
- 11.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Gill PS, Wilcox CS. NADPH oxidases in the kidney. AntioxidRedoxSignal. 2006;8(9–10):1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 13.Battersby AC, Cale CM, Goldblatt D, Gennery AR. Clinical Manifestations of Disease in X-Linked Carriers of Chronic Granulomatous Disease. Journal of Clinical Immunology. 2013;33(8):1276–1284. doi: 10.1007/s10875-013-9939-5. [DOI] [PubMed] [Google Scholar]
- 14.Djamali A, Vidyasagar A, Adulla M, Hullett D, Reese S. Nox-2 Is a Modulator of Fibrogenesis in Kidney Allografts. Am J Transplant. 2009;9(1):74–82. doi: 10.1111/j.1600-6143.2008.02463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djamali A, Reese S, Hafez O, Vidyasagar A, Jacobson L, Swain W, et al. Nox2 is a mediator of chronic CsA nephrotoxicity. Am J Transplant. 2012;12(8):1997–2007. doi: 10.1111/j.1600-6143.2012.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Hoffman BB, Lee JS, Kasama Y, Jim B, Kopp JB, et al. Cultured tubule cells from TGF-beta1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney Int. 2004;65(4):1191–1204. doi: 10.1111/j.1523-1755.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- 17.Bell CL, Tenenhouse HS, Scriver CR. Initiation and characterization of primary mouse kidney epithelial cultures. In vitro cellular & developmental biology : journal of the Tissue Culture Association. 1988;24(7):683–695. doi: 10.1007/BF02623606. [DOI] [PubMed] [Google Scholar]
- 18.Vidyasagar A, Reese SR, Hafez O, Huang LJ, Swain WF, Jacobson LM, et al. Tubular expression of heat-shock protein 27 inhibits fibrogenesis in obstructive nephropathy. Kidney international. 2013;83(1):84–92. doi: 10.1038/ki.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidyasagar A, Reese S, Acun Z, Hullett D, Djamali A. HSP27 is involved in the pathogenesis of kidney tubulointerstitial fibrosis. Am J Physiol Renal Physiol. 2008;295(3):F707–F716. doi: 10.1152/ajprenal.90240.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiernan JA. Histological and histochemical methods : theory and practice. 3rd ed. London England ; New York: Arnold; 1999. [Google Scholar]
- 21.Irish WD, Ilsley JN, Schnitzler MA, Feng S, Brennan DC. A risk prediction model for delayed graft function in the current era of deceased donor renal transplantation. Am J Transplant. 2010;10(10):2279–2286. doi: 10.1111/j.1600-6143.2010.03179.x. [DOI] [PubMed] [Google Scholar]
- 22.Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. 2011;11(11):2279–2296. doi: 10.1111/j.1600-6143.2011.03754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solhjou Z, Athar H, Xu Q, Abdi R. Emerging therapies targeting intra-organ inflammation in transplantation. Am J Transplant. 2015;15(2):305–311. doi: 10.1111/ajt.13073. [DOI] [PubMed] [Google Scholar]
- 24.Decleves AE, Sharma K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol. 2014;10(5):257–267. doi: 10.1038/nrneph.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Jung KJ, Park KM. Reactive oxygen species differently regulate renal tubular epithelial and interstitial cell proliferation after ischemia and reperfusion injury. Am J Physiol Renal Physiol. 2010;298(5):F1118–F1129. doi: 10.1152/ajprenal.00701.2009. [DOI] [PubMed] [Google Scholar]
- 26.Jang HS, Kim JI, Jung KJ, Kim J, Han KH, Park KM. Bone marrow-derived cells play a major role in kidney fibrosis via proliferation and differentiation in the infiltrated site. Biochim Biophys Acta. 2013;1832(6):817–825. doi: 10.1016/j.bbadis.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Jang HS, Kim JI, Han SJ, Park KM. Recruitment and subsequent proliferation of bone marrow-derived cells in the postischemic kidney are important to the progression of fibrosis. Am J Physiol Renal Physiol. 2014;306(12):F1451–F1461. doi: 10.1152/ajprenal.00017.2014. [DOI] [PubMed] [Google Scholar]
- 28.Nlandu Khodo S, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E, et al. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol. 2012;23(12):1967–1976. doi: 10.1681/ASN.2012040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhan CD, Sindhu RK, Vaziri ND. Up-regulation of kidney NAD(P)H oxidase and calcineurin in SHR: reversal by lifelong antioxidant supplementation. Kidney Int. 2004;65(1):219–227. doi: 10.1111/j.1523-1755.2004.00372.x. [DOI] [PubMed] [Google Scholar]
- 30.Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003;63(1):179–185. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- 31.Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, et al. Classical Inhibitors of NOX NAD(P)H Oxidases Are Not Specific. Curr Drug Metab. 2008;9(8):686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 32.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51(2):211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 33.Djamali A, Vidyasagar A, Yagci G, Huang LJ, Reese S. Mycophenolic Acid May Delay Allograft Fibrosis by Inhibiting Transforming Growth Factor-beta1-Induced Activation of Nox-2 Through the Nuclear Factor-kappaB Pathway. Transplantation. 2010 doi: 10.1097/TP.0b013e3181e6ae0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24(5):1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, et al. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26(1):140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Lohneysen K, Noack D, Wood MR, Friedman JS, Knaus UG. Structural insights into Nox4 and Nox2: motifs involved in function and cellular localization. Mol Cell Biol. 30(4):961–975. doi: 10.1128/MCB.01393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du T, Zou X, Cheng J, Wu S, Zhong L, Ju G, et al. Human Wharton's jelly-derived mesenchymal stromal cells reduce renal fibrosis through induction of native and foreign hepatocyte growth factor synthesis in injured tubular epithelial cells. Stem cell research & therapy. 2013;4(3):59. doi: 10.1186/scrt215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.