

Abstract
The Ca2+/calmodulin(CaM)-dependent protein kinase II (CaMKII) forms 12meric holoenzymes. These holoenzymes cluster into larger aggregates within neurons under ischemic conditions and in vitro when ischemic conditions are mimicking. This aggregation is thought to be mediated by interaction between the regulatory domain of one kinase subunit with the T-site of another kinase subunit in a different holoenzyme, an interaction that requires stimulation by Ca2+/CaM and nucleotide for its induction. This model makes several predictions that were verified here: Aggregation in vitro was reduced by the CaMKII inhibitors tatCN21 and tatCN19o (which block the T-site) as well as by KN93 (which is CaM-competitive). Notably, these and previously tested manipulations that block CaMKII activation all reduced aggregation, suggesting an alternative mechanism that instead requires kinase activity. However, experiments with the nucleotide-competitive broad-spectrum kinase inhibitors staurosporin and H7 showed that this is not the case. In vitro, staurosporine and H7 enabled CaMKII aggregation even in the absence of nucleotide. Within rat hippocampal neurons, an intra-body enabled live monitoring of endogenous CaMKII aggregation. This aggregation was blocked by tatCN21, but not by staurosporine, even though both effectively inhibit CaMKII activity. These results support the mechanistic model for CaMKII aggregation and show that kinase activity is not required.
Keywords: CaMKII, Aggregation, Ischemia, Glutamate, Hippocampus, FingR
Introduction
CaMKII is a major regulator of glutamatergic synaptic transmission in the brain, including that of long-term potentiation (LTP) and depression (LTD) (Coultrap & Bayer 2012b, Lisman et al. 2012, Hell 2014, Coultrap et al. 2014), two opposing forms of synaptic plasticity that are thought to underlie higher brain functions such as cognition, learning, and memory (Martin et al. 2000, Malenka & Bear 2004, Lee & Silva 2009). Glutamate is the major physiological excitatory neurotransmitter in the mammalian brain, but excessive glutamate release during ischemic conditions is also the trigger for the subsequent excitotoxic death of neurons (Choi et al. 1988, Doyle et al. 2008, Hara & Snyder 2007, Aarts & Tymianski 2004). Interestingly, this pathological glutamate signaling is also mediated by CaMKII and can be alleviated by CaMKII inhibition (Vest et al. 2010, Ashpole & Hudmon 2011, Coultrap et al. 2011). While LTP- and LTD-related glutamate stimuli trigger CaMKII translocation to excitatory and inhibitory synapses, respectively (Shen & Meyer 1999, Bayer et al. 2001, Zhang et al. 2008, Rose et al. 2009, Marsden et al. 2010, Coultrap & Bayer 2012b), excitotoxic glutamate stimuli trigger additional extrasynaptic aggregation of many CaMKII holoenzymes – which are themselves large 12meric complexes (Kanaseki et al. 1991, Chao et al. 2011, Coultrap & Bayer 2012b) – into larger clusters (Suzuki et al. 1994, Dosemeci et al. 2000, Tao-Cheng et al. 2002, Hudmon et al. 2005, Vest et al. 2009). A similar aggregation of purified CaMKII holoenzymes can also be induced in vitro by mimicking ischemic conditions biochemically, i.e. by incubating the kinase at a pH of 6.8 or lower in the presence of Ca2+ (and CaM) and ATP at low concentration or ADP at high concentration (Hudmon et al. 1996, Vest et al. 2009).
CaMKII mutations that prevent Ca2+/CaM stimulation and the K42M kinase dead mutation block the glutamate-induced holoenzyme aggregation within neurons (Vest et al. 2009, Hudmon et al. 2005), apparently indicating a requirement for CaMKII activity. This seems to be further corroborated by the inhibition of aggregation by the CaMKII inhibitors KN93, tatCN21, and tatCN19o that was seen in our current study. However, the current model for the molecular mechanism underlying aggregation (Fig. 1) (Vest et al. 2009) predicts activity-independent reasons for the block of aggregation by inactivating mutations and by inhibitors. In the basal state of CaMKII, the substrate binding S-site and the nearby T-site are blocked by the regulatory domain (Fig. 1) (Bayer et al. 2001, Chao et al. 2010, Coultrap & Bayer 2012b). Aggregation is thought to be mediated by the interaction between the regulatory domain of one kinase subunit with the T-site of another kinase subunit within a different holoenzyme (Fig. 1). Thus, aggregation requires stimulation by Ca2+/CaM (Hudmon et al. 1996, Vest et al. 2009), which can explain the block of aggregation by the A302R and the T305/306D mutations (Hudmon et al. 2005) and by the inhibitor KN93, which all prevent CaMKII stimulation by Ca2+/CaM. The inhibitors tatCN21 and tatCN19o (Vest et al. 2007, Coultrap & Bayer 2011) bind to the T-site of CaMKII, thereby explaining the block of aggregation by these inhibitors, as well as by the T-site mutant I205K (Hudmon et al. 2005). Additionally, aggregation requires occupation of the nucleotide binding pocket on CaMKII (Vest et al. 2009), which can explain the block of aggregation by the K42M mutation that prevents nucleotide binding (Vest et al. 2009).
Fig. 1.
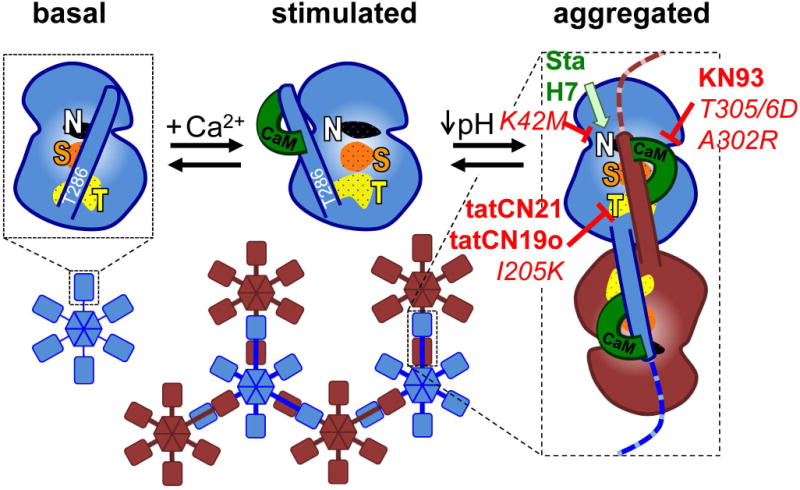
The current mechanistic model for aggregation of CaMKII holoenzymes into larger clusters. In the basal state (shown for an individual kinase subunit without depiction of the C-terminal association domain), the regulatory domain blocks the substrate-binding S-site (S, orange) and the neighboring T-site (named for its interaction with the T286 region of the regulatory domain; T, yellow); the nucleotide-binding pocket (N, white) is also indicated. Ca2+/CaM binding replaces the regulatory domain to allow access to the S- and T-sites. Then, the T-site can interact with binding partners such as GluN2B. T-site interaction with the regulatory domain of another kinase subunit additionally requires a drop in pH to ~6.8 or lower and then causes aggregation of multiple 12meric holoenzymes into large aggregates (shown here as hexamers and in two different colors for better visualization of the aggregates). Both aggregation and GluN2B binding additionally require occupation of the nucleotide binding pocket. Both aggregation and GluN2B binding is prevented by inhibitory mutations that prevent nucleotide binding (K42M) or Ca2+/CaM binding (T305/306D) as well as by inhibitors that block the T-site (tatCN21, tatCN19o) or are competitive with Ca2+/CaM (KN93). By contrast, nucleotide competitive inhibitors (staurosporine, H7) block enzymatic kinase activity but can replace the nucleotide function for aggregation and GluN2B binding. The differential inhibitor effects on aggregation were elucidated by the results of this study.
In order to further test the current mechanistic model for CaMKII holoenzyme aggregation and to determine the requirement for CaMKII activity in aggregation within neurons, we used a similar strategy as we had previously employed to address similar questions about the CaMKII interaction with the NMDA-receptor subunit GluN2B (Barcomb et al. 2013), an interaction that also requires the CaMKII T-site and is stimulated by binding of Ca2+/CaM and nucleotide to CaMKII (Bayer et al. 2001, Bayer et al. 2006, O’Leary et al. 2011). As predicted by the current model (Fig. 1), and as briefly delineated above, CaMKII aggregation in vitro was blocked by the CaMKII inhibitors KN93, tatCN21, and tatCN19o. However, inhibiting CaMKII activity by the ATP-competitive broad-spectrum kinase inhibitors staurosporine or H7 did not block the in vitro aggregation. Thus, similar to the CaMKII/GluN2B interaction (Barcomb et al. 2013), occupation of the nucleotide binding pocket by staurosporine (Omura et al. 1977, Tamaoki et al. 1986, Nakano et al. 1987, Yanagihara et al. 1991) or H7 (Hidaka et al. 1984, Malinow et al. 1989) substituted for the nucleotide requirement (rather than interfering with it). This result enabled testing the requirement of CaMKII activity in aggregation within neurons. While aggregation was blocked by tatCN21 (as predicted based on T-site interference), it was not inhibited by staurosporine, demonstrating that activity of CaMKII (or the other kinases inhibited by staurosporine) is not required for aggregation. These results also highlight the need for detailed mechanistic knowledge of inhibitors and inactivating mutations in order to correctly infer the requirement of enzymatic activities in biological processes. In this case, the effect of several different inhibitors or inhibiting mutations on CaMKII aggregation was through structural mechanisms unrelated to the inhibition of enzymatic activity.
Materials and methods
Materials
CaMKIIα and CaM were purified after baculovirus/Sf9 cell expression or bacterial expression as previously described (Coultrap et al. 2010, Coultrap & Bayer 2012a). The vector for the CaMKIIα FingR was described previously (Mora et al. 2013). Cell culture reagents and chemicals were obtained from Invitrogen and Sigma, respectively, unless indicated otherwise. H7 (1-(5-isoquinolinylsulfonyl)-2-methylpiperazine) and staurosporine were obtained from Tocris. TatCN21, TatCN19o and TatSCR peptides were custom ordered through CHI Scientific.
CaMKII aggregation in vitro
Aggregation of purified CaMKIIα (0.5 μM) was induced in 30–35 mM PIPES at pH 6.5–6.8, 20 mM KCl, 10 mM MgCl2, 0.1 mg/ml BSA, 0.1% Tween, 0.5 mM DTT, 0.5 mM CaCl2, 1.5–2 μM CaM and 0.1–1 mM ADP (unless indicated otherwise), essentially as described previously (Vest et al. 2009). Various inhibitors were present in the mixture as indicated, prior to induction of aggregation by the addition of nucleotide (or the nucleotide-competitive inhibitors H7 or staurosporin). After 5 min at room temperature, soluble and aggregated CaMKII were separated by centrifugation at 14,000xg at 4°C for 30 min. Western blot analysis of the resulting pellet and supernatant was performed using a monoclonal CaMKIIα antibody (CBα2; Invitrogen) and quantified as described previously (Vest et al. 2007, Coultrap & Bayer 2012a, Goodell et al. 2014, Coultrap et al. 2014). The quantified amount of CaMKII in the pellet was normalized, with the aggregation in presence of ADP (≥100 μM) without inhibitors in the same experiment set to 100.
Preparation, transfection, and live imaging of primary neuronal cultures
Pregnant Sprague-Dawley rats were supplied by Charles River Labs for the preparation of primary hippocampal cultures from P0–P1 neonatal rat pups as described previously (Barcomb et al. 2014, Vest et al. 2007). All animal treatment was in accordance with the University of Colorado Denver Institutional Animal Care and Use Committee. At 12 DIV neurons were transfected with the CaMKII FingR intrabody using Lipofectamine 2000. The following day the neurons were imaged using a Zeiss Axiovert 200M microscope with climate controlled chamber at 32°C in a HEPES buffered solution (0.87X Hank’s Buffered Saline Solution, 25 M HEPES pH 7.4, 2 mM glucose, 2 mM CaCl2 and 1 mM MgCl2) containing tatCN21 (5 μM), tatCN19o (5 μM), staurosporine (2 μM), or no inhibitor. Z-stack images were collected every 10 seconds for 3 minutes, with glutamate/glycine (100 μM/10 μM) stimulation at 30 seconds. Images were analyzed blind at 2 minutes after stimulation for the number of puncta, which were defined as discrete objects greater than 2 pixels in size with intensity greater than 3 standard deviations from the mean somal intensity.
Statistical analysis
All quantifications are shown as mean±SEM and were tested for statistically significant differences by ANOVA followed by Tukey’s posthoc analysis (ns: p>0.05; *: p<0.05; **: p<0.01; ***: p<0.001) using GraphPad Prism.
Results
CaMKII aggregation in vitro is blocked by the inhibitors KN93, tatCN21, and tatCN19o
Aggregation of CaMKII holoenzymes into larger clusters in vitro was induced by the addition of nucleotide (1 mM ADP) at pH ≤6.8 in the presence of Ca2+/CaM, and detected by a differential centrifugation assay followed by Western blot analysis of the supernatant (containing soluble CaMKII holoenzymes) and the pellet (containing the aggregated holoenzymes). As predicted by the molecular model for CaMKII aggregation (Vest et al. 2007) (see Fig. 1), the Ca2+/CaM-competitive CaMKII inhibitor KN93 (10 μM) significantly reduced aggregate formation (Fig. 2). By contrast, the inactive analogue KN92 (10 μM) had no effect (Fig. 2). As also predicted by the model, the T-site binding CaMKII inhibitor peptides tatCN19o (5 μM) or tatCN21 (5 μM) both significantly reduced aggregation (Fig. 2). By contrast, the inactive scrambled control peptide tatCtrl (5 μM) had no effect.
Fig. 2.
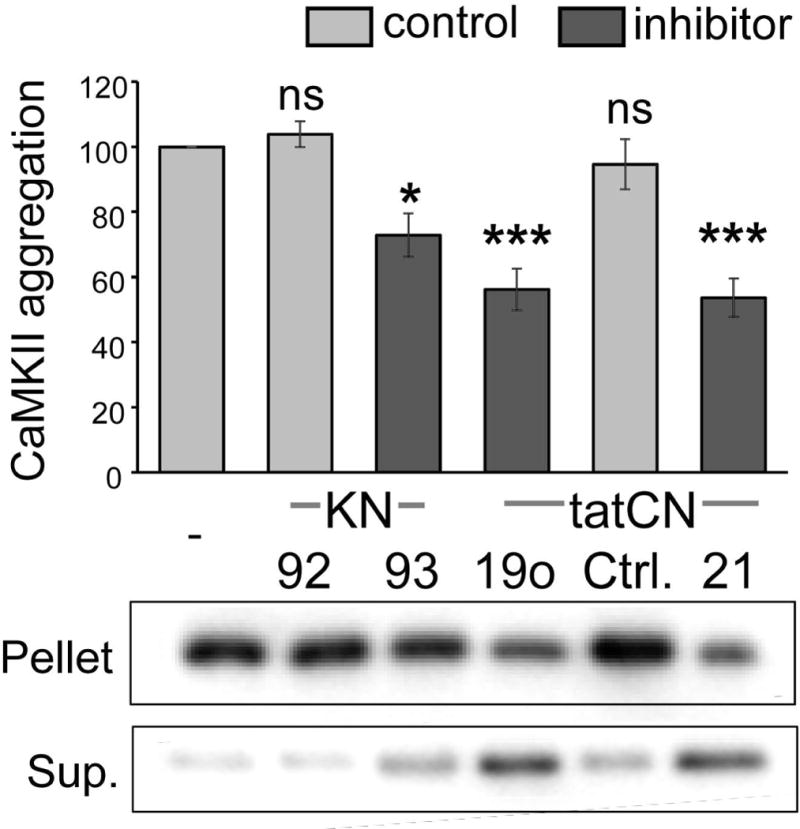
CaMKII aggregation in vitro is reduced by the CaMKII inhibitors KN93, tatCN19o and tatCN21. CaMKII aggregation was induced at pH 6.5–6.8 by the addition of 1 mM ADP in the presence of Ca2+/CaM and 10 μM KN93, KN92, 5 μM tatCN19o, tatCtrl, or tatCN21, as indicated (with 10 μM KN compounds or 5 μM tat peptides). Aggregates were separated by centrifugation from soluble kinase, and both pellet and supernatant (Sup.) were analyzed for CaMKII content by Western blot. Aggregation was normalized to ADP-only control. CaMKII inhibitor (dark grey) versus control substance (light grey) conditions are indicated. One-Way ANOVA with post-hoc Tukey’s test indicated that the inhibitors KN93, tatCN19o and tatCN21 significantly reduced aggregation (* p<0.05, *** p<0.001, ns = non-significant as compared to ADP-only control; n=5–6).
These results further corroborate the current molecular model for CaMKII aggregation (see Fig. 1). However, the observed inhibition of CaMKII aggregation by KN93, tatCN21 and tatCN19o also makes these inhibitors unsuitable for testing whether or not CaMKII activity is required for aggregation within neurons.
The nucleotide-competitive inhibitors staurosporine or H7 can substitute for nucleotide in the induction of CaMKII aggregation in vitro
The proposed mechanisms for CaMKII aggregation and for CaMKII binding to GluN2B share several similarities, and both are promoted by nucleotide binding to the kinase (Vest et al. 2007, O’Leary et al. 2011). Within cells, both CaMKII aggregation and GluN2B binding are blocked by the CaMKII K42M mutation that prevents nucleotide binding (Vest et al. 2009, O’Leary et al. 2011). However, the nucleotide-competitive inhibitors staurosporine and H7 do not block the CaMKII/GluN2B interaction; instead, occupation of the CaMKII nucleotide binding pocket by these inhibitors can substitute for nucleotide binding (O’Leary et al. 2011). Thus, we here tested if these nucleotide-competitive inhibitors can also substitute for nucleotides in the regulation of CaMKII aggregation. Indeed, while no CaMKII aggregation was observed in the absence of nucleotide, addition of ADP (≥100 μM), H7 (700 μM), or staurosporine (2 μM) induced robust aggregation (Fig. 3). Consistent with previous reports, ADP induced aggregation at high concentrations (100 μM), while lower concentrations (10 μM) were not sufficient (Fig. 3). For staurosporin, 2 μM was sufficient to induce aggregation (Fig. 3), consistent with its higher affinity for the nucleotide binding pocket. Thus, CaMKII aggregation is enabled by occupation of the nucleotide binding pocket either by nucleotide or by nucleotide-competitive inhibitors. Importantly, identification of these kinase inhibitors that do not interfere with CaMKII aggregation allows for testing of the requirement of enzymatic kinase activity in ischemia-related CaMKII aggregation within neurons.
Fig. 3.
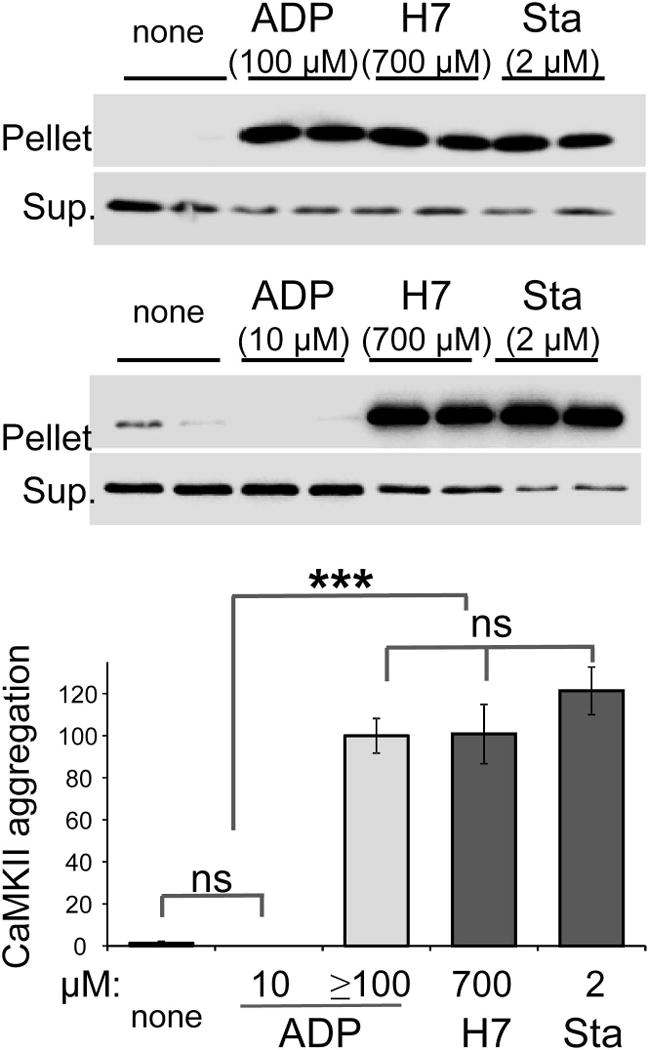
The nucleotide-competitive inhibitors staurosporine (Sta) and H7 can substitute for nucleotide in inducing CaMKII aggregation in vitro. CaMKII aggregation was induced at pH 6.5–6.8 in the presence of Ca2+/CaM by addition of ADP, H7, or staurosporine at the concentrations indicated. Without addition of inhibitor or nucleotide (none), no aggregation was seen. Aggregates were separated by centrifugation from soluble kinase, and both pellet and supernatant (Sup.) was analyzed for CaMKII content by Western blot. Aggregation was normalized to ADP positive control. One-Way ANOVA with post-hoc Tukey’s test showed that ≥100 μM ADP (n=3), 700 μM H7 (n=4), 2 μM Sta (n=5) equally and significantly induced CaMKII aggregation, compared to either no nucleotide (n=4) or 10 μM ADP (n=2) (*** p<0.001, ns = non-significant).
Live-imaging of the aggregation of endogenous CaMKII within neurons
GFP-labelled FingRs (Fibronectin intrabodiesgenerated with mRNA display) are powerful new tools for live imaging of endogenous proteins within cells (Gross et al. 2013, Mora et al. 2013). When expressed in cells, these labeled intrabodies bind specifically to the endogenous protein, allowing live molecular imaging without any potential distortion that can be caused by the overexpression of a protein that is directly tagged with a fluorescent protein (Gross et al. 2013, Mora et al. 2013). However, like direct GFP-labeling, FingR binding has the potential to alter specific functions of the labeled protein. Thus, we decided to test here whether or not a CaMKII FingR (Mora et al. 2013) interferes with the glutamate-induced CaMKII aggregation within hippocampal neurons. As previously observed by immuno-EM for endogenous CaMKII (Dosemeci et al. 2000, Tao-Cheng et al. 2002) and by fluorescence microscopy for overexpressed GFP-CaMKII (Vest et al. 2009, Hudmon et al. 2005), treatment with 100 μM glutamate plus 10 μM glycine induced robust CaMKII aggregation in hippocampal neurons that was readily detectable with the expressed FingR (Fig. 4 and Supplemental Movie 1). The extrasynaptic nature of CaMKII aggregates was determined based on serial planes of deconvoluted z-stack images (each plane 0.3 μm apart within a 1.8 μm stack), which demonstrated aggregates that are not associated with the plasma membrane (Fig. 4). Thus, the CaMKII FingR does not prevent extrasynaptic CaMKII aggregation and can be utilized for monitoring this aggregation of endogenous CaMKII within live neurons.
Fig. 4.
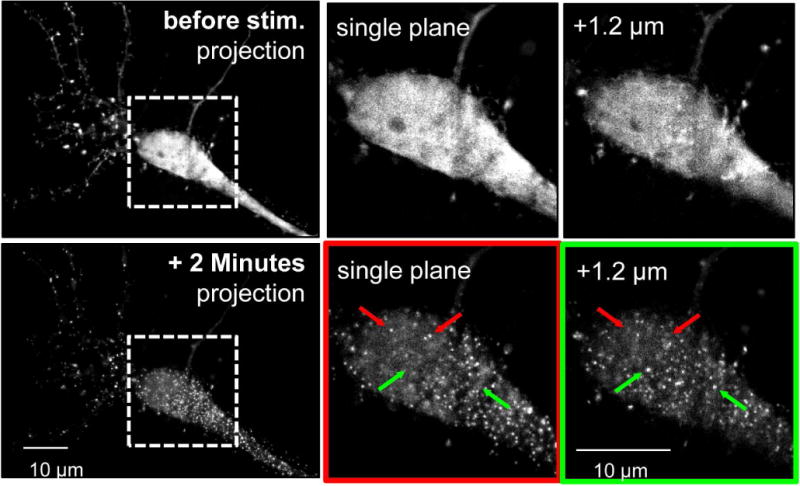
A FingR that enables live imaging of aggregation of endogenous CaMKII within neurons. Primary hippocampal neuron cultures were transfected with a CaMKII FingR at 12 DIV, allowing for visualization of endogenous CaMKII (at 13 DIV). (Top) Basally CaMKII is not aggregated in the soma, as seen in a representative neuron. Images of neurons were collected in z-stacks over 1.8 μm with 0.3 μm between planes. Somal puncta are not seen in a flattened image of all of the planes or in individual planes; top and bottom planes of the boxed in region are shown on the right. (Bottom) Aggregation is clearly seen 2 minutes after glutamate/glycine stimulation (stim.) in the same neuron depicted in the top panel. Arrows in the right panels identify puncta that are present in either the top plane (red) or the bottom plane (green) but not both. The presence of unique puncta in individual planes throughout the z-stacks shows that they are cytoplasmic rather than membrane associated.
CaMKII aggregation within neurons does not require kinase activity
Both staurosporine (2 μM) and tatCN21 (5 μM) effectively block CaMKII activity within cells (Vest et al. 2007, Barcomb et al. 2013); H7 can be similarly effective, but only at much higher concentrations (≥700 μM) (Barcomb et al. 2013). Thus, we decided to utilize staurosporine (which did not inhibit CaMKII aggregation in vitro; see Fig. 3) to test whether or not CaMKII activity is required for aggregation within neurons (Fig. 5). Glutamate-induced aggregation of endogenous CaMKII was monitored live in cultured hippocampal neurons using the FingR method described in the previous section. First, we tested if CaMKII aggregation in neurons can be inhibited by pharmacological means in principle. Indeed, 5 μM tatCN21 or tatCN19o (the T-site binding CaMKII inhibitors that blocked CaMKII aggregation in vitro; see Fig. 2) significantly reduced aggregation of endogenous CaMKII within the neurons (Fig. 5). By contrast, inhibiting CaMKII with 2 μM staurosporine had no significant effect on the aggregation, neither for endogenous CaMKII (Fig. 5) nor for overexpressed GFP-CaMKII (Supplemental Fig. 1). These results further validate the current mechanistic model for CaMKII aggregation, and demonstrate that CaMKII aggregation within neurons does not require enzymatic activity of CaMKII (or of any other kinase inhibited by staurosporine).
Fig. 5.
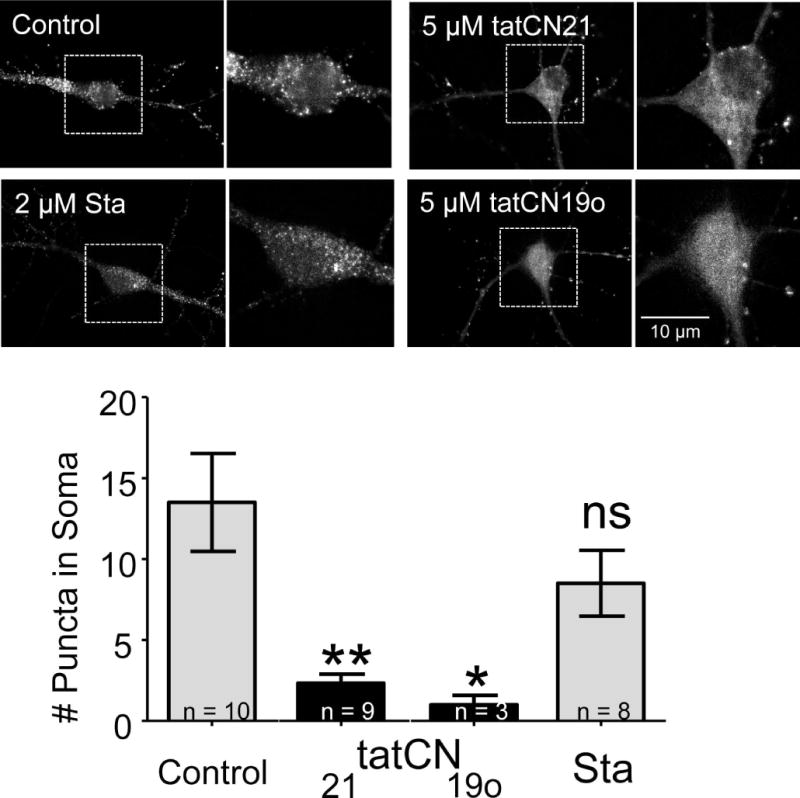
CaMKII aggregation within neurons does not require enzymatic kinase activity. CaMKII FingR transfected neurons were imaged in the presence of tatCN21 (5 μM; n=9), tatCN19o (5 μM; n=3), staurosporine (Sta; 2 μM; n=8), or no inhibitor (control; n=10). The top panels show representative images of cells in all four conditions 2 minutes after glutamate/glycine stimulation. Puncta – indicating aggregation – can clearly be seen in the control and staurosporine conditions, but not in either of the tat-peptide conditions. The images were quantified by counting the number of somal puncta, as defined by objects greater than 2 pixels in size whose intensity was greater than 3 standard deviations from the mean somal intensity at 2 minutes. This value was significantly less than control for tatCN19o and tatCN21, as determined by a One-Way ANOVA with post-hoc Tukey’s test (* p<0.05, ** p<0.01, ns = non-significant as compared to control).
Discussion
Ischemia-related CaMKII aggregation within neurons is prevented by mutations (K42M or A302R and T305/306D) that prevent CaMKII activity by two independent mechanisms (Vest et al. 2009, Hudmon et al. 2005). However, the current study demonstrates that this aggregation does not require enzymatic activity of CaMKII. While the T-site binding inhibitors tatCN21 and tatCN19o (Vest et al. 2007, Coultrap & Bayer 2011) also inhibited CaMKII aggregation both in vitro and within neurons (as predicted by the mechanistic model for aggregation shown in Fig. 1), CaMKII inhibition with staurosporine did not affect aggregation, neither in vitro nor within neurons. While staurosporine is a broad-spectrum kinase inhibitor that is not restricted to CaMKII, these results show that the enzymatic activities of either CaMKII or any of the other kinases inhibited by staurosporine (including PKA and PKC (Meggio et al. 1995)) are not required for CaMKII aggregation. Notably, the staurosporine concentration used here (2 μM) was ~230x the Ki for CaMKII inhibition and has been demonstrated to effectively inhibit CaMKII activity within cells (Barcomb et al. 2013).
It should be pointed out that high concentrations of tatCN21 by itself can induce a different, artificial form of CaMKII aggregation in neurons that is distinguished by an unusual association with polyribosomes (Tao-Cheng et al. 2013). However, these polyribosome aggregates were prevalent only at higher concentrations of tatCN21 (20 μM) and were seen only minimally, or not at all, at the lower concentration used here (5 μM) to inhibit formation of the normal self-aggregates.
Even though the current mechanistic model for CaMKII aggregation (see Fig. 1) predicts that kinase activity is not required, it does stipulate the requirement for nucleotide binding to CaMKII. The latter is consistent with the failure of the nucleotide binding-impaired CaMKII K42M mutant to aggregate (Vest et al. 2007), but it is in apparent conflict with the aggregation observed here in the presence of nucleotide binding-competitive inhibitors such as staurosporine. However, a similar situation has been observed previously for the CaMKII/GluN2B interaction that is also positively regulated by nucleotides (Barcomb et al. 2013): While the occupation of the nucleotide binding pocket of CaMKII by staurosporine instead of a nucleotide prevents enzymatic kinase activity, it can substitute for nucleotides in enhancing GluN2B binding or in enabling aggregation. Indeed, our in vitro experiments demonstrate that CaMKII aggregation was enabled alternatively by the addition of either nucleotide or the nucleotide-competitive inhibitors staurosporine or H7. By contrast, and as predicted by the model shown in Fig.1, the Ca2+/CaM-competitive CaMKII inhibitor KN93 (Vest et al. 2010, Tokumitsu et al. 1990, Sumi et al. 1991) and the T-site binding CaMKII inhibitors tatCN19o and tatCN21 (Vest et al. 2007, Coultrap & Bayer 2011) were found here to inhibit CaMKII aggregation in vitro, even under conditions where enzymatic kinase activity was already disabled by substitution of ATP with ADP. Thus, our results further validate the current mechanistic model for CaMKII aggregation, and provide additional evidence that the CaMKII aggregation in vitro and within neurons are regulated by the same mechanisms.
Our results also provide the first live observation of aggregation of endogenous CaMKII within neurons. Previously, extrasynaptic CaMKII aggregation has been observed either live, but only for overexpressed GFP-CaMKII (Vest et al. 2009, Hudmon et al. 2005), or for endogenous CaMKII, but only by immunostaining in fixed neurons (Dosemeci et al. 2000, Tao-Cheng et al. 2002). Live imaging of endogenous protein within cells is enabled by the expression of GFP-labeled intrabodies (Gross et al. 2013, Mora et al. 2013). The use of such intrabodies instead of expressing directly GFP-labeled protein avoids functional alteration that can be caused by overexpression. For instance, PSD95 and gephyrin intrabodies have been used to label excitatory and inhibitory synapses, respectively, without the functional alterations of these synapses seen after overexpression of the corresponding directly GFP-labeled proteins (Gross et al. 2013). However, like direct GFP-labelling, indirect labeling with intrabodies has the potential to alter specific functions of the labeled protein. Thus, in order to monitor cellular processes with intrabodies, it has to be established first that the intrabodies do not interfere with these processes. The CaMKII FingR intrabody utilized here was previously shown to allow normal CaMKII translocation to excitatory synapses in response to LTP-related glutamate stimuli (Mora et al. 2013). Our results show that this FingR also allows normal extrasynaptic CaMKII aggregation in response to excitotoxic glutamate stimuli.
The current study demonstrates another aspect of the interaction, the requirement of nucleotide-binding pocket occupation but not of enzymatic activity, and further confirms the mechanistic similarities between CaMKII aggregation and CaMKII/GluN2B binding. This provides further validation for the current mechanistic model for CaMKII aggregation (illustrated in Fig. 1). Additionally, this study establishes a technique for monitoring the self-aggregation of endogenous CaMKII within live cells, which can be further exploited in the future.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health (NIH) grants F31NS083298 (to K.B.), F31NS092265 (to D.J.G), T32HD041697 (D.J.G.), P30NS048154 (UCD center grant), R01NS081248, and R01NS080851 (to K.U.B).
Footnotes
Conflict of Interest Statement: The University of Colorado holds patent rights for inhibitors used in this study (US patent US 8,816,046 B2). K.U.B is the owner of Neurexus Therapeutics, LLC.
References
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4:137–147. doi: 10.2174/1566524043479202. [DOI] [PubMed] [Google Scholar]
- Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. 2011;46:720–730. doi: 10.1016/j.mcn.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Barcomb K, Buard I, Coultrap SJ, Kulbe JR, O’Leary H, Benke TA, Bayer KU. Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J. 2014;28:3810–3819. doi: 10.1096/fj.14-250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Coultrap SJ, Bayer KU. Enzymatic Activity of CaMKII Is Not Required for Its Interaction with the Glutamate Receptor Subunit GluN2B. Mol Pharmacol. 2013;84:834–843. doi: 10.1124/mol.113.089045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, Kuriyan J. Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat Struct Mol Biol. 2010;17:264–272. doi: 10.1038/nsmb.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell. 2011;146:732–745. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. Improving a Natural CaMKII Inhibitor by Random and Rational Design. PLoS One. 2011;6:e25245. doi: 10.1371/journal.pone.0025245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) In: Mukai H, editor. Neuromethods: Protein Kinase Technologies. Springer; 2012a. pp. 49–72. [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012b;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Buard I, Kulbe JR, Dell’Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J Biol Chem. 2010;285:17930–17937. doi: 10.1074/jbc.M109.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O’Leary H, Sanderson JL, Roche KW, Dell’Acqua ML, Bayer KU. Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Reports. 2014;6:431–437. doi: 10.1016/j.celrep.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU. CaMKII in cerebral ischemia. Acta Pharmacol Sin. 2011;32:861–872. doi: 10.1038/aps.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS, Petersen J, Tao-Cheng JH. A novel particulate form of Ca(2+)/calmodulin-dependent [correction of Ca(2+)/CaMKII-dependent] protein kinase II in neurons. J Neurosci. 2000;20:3076–3084. doi: 10.1523/JNEUROSCI.20-09-03076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell DJ, Eliseeva TA, Coultrap SJ, Bayer KU. CaMKII binding to GluN2B is differentially affected by macromolecular crowding reagents. PLoS One. 2014;9:e96522. doi: 10.1371/journal.pone.0096522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GG, Junge JA, Mora RJ, et al. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron. 2013;78:971–985. doi: 10.1016/j.neuron.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–141. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Hell JW. CaMKII: claiming center stage in postsynaptic function and organization. Neuron. 2014;81:249–265. doi: 10.1016/j.neuron.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Aronowski J, Kolb SJ, Waxham MN. Inactivation and self-association of Ca2+/calmodulin-dependent protein kinase II during autophosphorylation. J Biol Chem. 1996;271:8800–8808. doi: 10.1074/jbc.271.15.8800. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaseki T, Ikeuchi Y, Sugiura H, Yamauchi T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J Cell Biol. 1991;115:1049–1060. doi: 10.1083/jcb.115.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc Natl Acad Sci U S A. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Meggio F, Donella Deana A, Ruzzene M, et al. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur J Biochem. 1995;234:317–322. doi: 10.1111/j.1432-1033.1995.317_c.x. [DOI] [PubMed] [Google Scholar]
- Mora RJ, Roberts RW, Arnold DB. Recombinant probes reveal dynamic localization of CaMKIIalpha within somata of cortical neurons. J Neurosci. 2013;33:14579–14590. doi: 10.1523/JNEUROSCI.2108-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Kobayashi E, Takahashi I, Tamaoki T, Kuzuu Y, Iba H. Staurosporine inhibits tyrosine-specific protein kinase activity of Rous sarcoma virus transforming protein p60. J Antibiot (Tokyo) 1987;40:706–708. doi: 10.7164/antibiotics.40.706. [DOI] [PubMed] [Google Scholar]
- O’Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, Bayer KU. Nucleotides and phosphorylation bi-directionally modulate Ca2+/calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-D-aspartate (NMDA) receptor subunit GluN2B. J Biol Chem. 2011;286:31272–31281. doi: 10.1074/jbc.M111.233668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchya H, Takahashi Y, Masuma R. A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot (Tokyo) 1977;30:275–282. doi: 10.7164/antibiotics.30.275. [DOI] [PubMed] [Google Scholar]
- Rose J, Jin SX, Craig AM. Heterosynaptic molecular dynamics: locally induced propagating synaptic accumulation of CaM kinase II. Neuron. 2009;61:351–358. doi: 10.1016/j.neuron.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K, Tanaka R, Tada T. Rapid translocation of cytosolic Ca2+/calmodulin-dependent protein kinase II into postsynaptic density after decapitation. J Neurochem. 1994;63:1529–1537. doi: 10.1046/j.1471-4159.1994.63041529.x. [DOI] [PubMed] [Google Scholar]
- Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Vinade L, Pozzo-Miller LD, Reese TS, Dosemeci A. Calcium/calmodulin-dependent protein kinase II clusters in adult rat hippocampal slices. Neuroscience. 2002;115:435–440. doi: 10.1016/s0306-4522(02)00451-7. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Yang Y, Bayer KU, Reese TS, Dosemeci A. Effects of CaMKII inhibitor tatCN21 on activity-dependent redistribution of CaMKII in hippocampal neurons. Neuroscience. 2013;244:188–196. doi: 10.1016/j.neuroscience.2013.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual Mechanism of a Natural CaMKII Inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Bayer KU. Differential regulation by ATP versus ADP further links CaMKII aggregation to ischemic conditions. FEBS Lett. 2009;583:3577–3581. doi: 10.1016/j.febslet.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J Biol Chem. 2010;285:20675–20682. doi: 10.1074/jbc.M109.088617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara N, Tachikawa E, Izumi F, Yasugawa S, Yamamoto H, Miyamoto E. Staurosporine: an effective inhibitor for Ca2+/calmodulin-dependent protein kinase II. J Neurochem. 1991;56:294–298. doi: 10.1111/j.1471-4159.1991.tb02595.x. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Holbro N, Oertner TG. Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proc Natl Acad Sci U S A. 2008;105:12039–12044. doi: 10.1073/pnas.0802940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.