

Abstract
In continuation of our ongoing search for bioactive compounds from microbial extracts, we performed antiproliferative and/or antimalarial assays on extracts of 806 microbial species isolated from Madagascan marine organisms, on 1317 species isolated from Madagascan soil samples and on a Streptomyces species (S.4) from a marine sponge collected from the Florida Keys. This work identified active extracts from four Streptomyces isolates (S.1, S.2, S.3 and S.4). The extracts of Streptomyces S.1 and S.2 showed antiproliferative activity against the A2780 ovarian cancer cell line, while those of S.3 and S4 displayed both antiproliferative and antimalarial activity. Bioassay-guided fractionation coupled with dereplication of the active extracts led to the identification and isolation of nonactin (1), monactin (2), dinactin (3), ±-nonactic acid (4), toyocamycin (5), piperafizine A (6) and a new dipeptide named xestostreptin (7). The structures of all isolated compounds 1–7 were elucidated by analyses of their NMR spectroscopic and mass spectrometric data, and were confirmed by comparison with the data reported in the literature. Compound 6 was crystallized and subjected to X-ray diffraction analysis to confirm its structure as piperafizine A (6). Compounds 1–3 displayed strong antiproliferative activity against A2780 ovarian cancer cells (IC50 values of 0.1, 0.13 and 0.2 μM, respectively), A2058 melanoma cells (IC50 values of 0.2, 0.02 and 0.02 μM, respectively), and H522-T1 non small-cell cancer lung cells (IC50 values of 0.1, 0.01 and 0.01 μM, respectively), while compounds 4 and 7 exhibited weak antiplasmodial activity against the Dd2 strain of Plasmodium falciparum, with IC50 values of 6.5 and 50 μM, respectively.
Keywords: Streptomyces, Antiproliferative, Antiplasmodial, Dereplication, Structure elucidation, Nuclear Magnetic Resonance
Graphical Abstract

Cancer and malaria are two of the major health threats to the world’s population. About 500 million malaria cases and 1 million deaths from malaria are reported each year.1 The best current antimalarial therapy is artemisinin combination therapy (ACT), but some cases of artemisinin resistance have been reported.2 Also, in the United States, cancer is the second leading cause of death after heart disease. There is thus a continuing need for new anticancer and antimalarial agents. New chemical entities with new pharmacophores are needed to tackle these public health problems.
Natural products have been the major contributor of antimalarial and anticancer drugs. Forty five percent of clinically used anticancer drugs3 and many antimalarial drugs are natural products or are derived from natural products. Among sources of natural products, Actinomycete bacteria produce a wide range of bioactive compounds with unique chemotypes and pharmacophores. With the aim of isolating bioactive molecules with new pharmacophores from Actinomycete species, we screened extracts of 806 microbial species isolated from Madagascan marine organisms, 1317 species isolated from Madagascan soil samples, and a Streptomyces species (S.4) from the sponge Xestospongia muta collected from the Florida Keys for antiproliferative activity against the A2780 ovarian cancer cell line and for antimalarial activity. A dereplication method leading to the isolation and structure determination of the known (1–6) and new (7) bioactive compounds as well as their biological activities are reported herein.
Although natural products are actively contributing to drug discovery by providing new pharmacophores and chemical entities, most of the major pharmaceutical companies have abandoned their natural product extracts screening program due to the high cost, the presence of known bioactive compounds that are often responsible for the activity, and the low yield. The problem of the re-isolation of known compounds can however be minimized by the use of appropriate dereplication methods. Several dereplication methods have been reported based on liquid chromatography coupled with mass spectrometry;4,5 the identification of compounds is often performed by comparison of their mass spectrometric data with those present in libraries of known bioactive compounds such as Antibase and Marinlit.
The evaluation of the potential of marine and soil microbial extracts as a source of antiproliferative and antimalarial compounds was one of several aims of the Madagascar ICBG program.6 In this study, a few milligrams of each microbial extract were received from the Centre National de Recherches sur l’Environnement (CNRE), Madagascar, and the Institute of Marine and Environmental Technology, University of Maryland Center for Environmental Science, Baltimore, for antiproliferative and antimalarial activity screenings. Among the more than 2,000 samples tested, 17 extracts showed antiproliferative activity with IC50 values of 20 μg/mL or less. The two most potent antiproliferative Streptomyces extracts, designated S.1 and S.2 with IC50 values of 2 μg/mL and 3.5 μg/mL, respectively, and two extracts designated S.3 and S.4 exhibiting antimalarial activity with IC50 values between 2.5 and 5 μg/mL, and 10 μg/mL, respectively, were selected for dereplication as described in Chart 1.
Chart 1.

A diagram of the dereplication method used during the present study
The two extracts S.1 and S.2 showing strong antiproliferative activity were subjected to liquid-liquid partition between water and ethyl acetate to remove polar compounds originating from the culture media. The active fractions ethyl acetate fractions were subjected to High Performance Liquid Chromatography (HPLC) and/or preparative TLC to obtain pure or semi-pure compounds for bioassay and NMR evaluation. The 1H NMR spectra of the most promising fractions were analyzed for the presence of known bioactive compounds by using the Dictionary of Natural Products (DNP) 1H-NMR and MarinLitdatabases.7
The 1H NMR spectrum of the ethyl acetate fraction of the extract obtained from S.1, which exhibited antiproliferative activity with an IC50 value of 2 μg/mL, showed the presence of mixtures of cyclic ionophores as substantiated by the triplet (δ 0.7 ppm) and doublet (δ 1~1.30 ppm) methyl signals in the upfield region of the 1H-NMR spectrum, and by oxygen-bearing methine multiplet signals (δ 3.70~4.99 ppm). Examination of 100 mg of extract obtained from scaled-up fermentation of the same strain led to the isolation of the antiproliferative compounds nonactin (1), dinactin (2), monactin (3) and ±-nonactic acid (4) from the most active fraction. The ethyl acetate fraction from Streptomyces sp. S.2 exhibited strong antiproliferative activity against A2780 cells (IC50 0.8 μg/mL). HPLC coupled with bioassay of this active fraction led to the isolation of a bioactive compound which was not retained on a Cogent reversed phase bidentate C-18 column (100A, 4μm, 50 x 4.6 mm). Its 1H NMR spectrum showed signals for α- and β-glucose and other signals ascribable to toyacamycin (5) the assignment of which was consistent with its UV and mass spectrometric data. The identity of 5 was confirmed by purification of a larger sample and by 1H and 13C NMR spectroscopy and mass spectrometric data as described in the experimental section.
Extract S.3 showed both antiproliferative (IC50 3.4 μg/mL) and antimalarial (IC50 ~4 μg/mL) activities. Open column chromatography of the ethyl acetate fraction on silica gel led to the isolation of piperafizine A (6), the structure of which was elucidated by analysis of its NMR spectroscopic data and x-ray diffraction (Fig. 1). Compound 6 is a condensation product of two phenylalanines, and has been isolated from Streptoverticillium aspergilloides (Q-576-2) and reported to be a potentiator of the cytotoxicity of vincristine.8, 9 Compound 6 showed antimalarial activity against P. falciparum with an IC50 value of 6.5 μM. This is the first report on the crystal structure and the antimalarial activity of 6.
Figure 1.

X-ray crystal structure of 6
The Streptomyces sp. isolated from the sponge X. muta(S.4) exhibited antimalarial activity with an IC50 value 10 μg/mL. Liquid-liquid partitioning followed by Sephadex LH-20 and HPLC of the ethyl acetate fraction yielded the new compound 7.
Compound 7 had the molecular formula C9H14N2O3 as determined by the high resolution ESIMS analysis, which exhibited a protonated-molecular ion peak at m/z 199.1068 (required for C9H15N2O3+, m/z 199.1077 [M+H]+).10 Its IR spectrum showed the presence of an absorption ascribable to an amide carbonyl function (1632 cm−1). The 1H NMR spectroscopic data recorded in CD3OD (Table 1) exhibited signals of: one secondary methyl (δ 1.26, d, J= 6.5 Hz, 3H), one methoxy (δ 3.32, s, 3H), one nitrogen bearing methyl (δ 3.10, s, 3H), two downfield methines (δ 3.76, qd, J= 6.5, 3.5Hz, H-7, 1H and 4.05, d, J= 3.5Hz, H-5, 1H) which were coupled to each other, and two proton signals of an exocyclic methylene (δ 4.88, s, H-and 5.41, s, each 1H). The HMBC spectroscopic data of 1 displayed correlations with 9 carbons (Table 1): two amide carbonyls (C-3 and C-6, at δ 160.2), three signals corresponding to an oxygenated carbon (δ 56.2), a nitrogen-bearing carbon (δ 35.6) and a secondary methyl group, two oxygen- and/or nitrogen-bearing methines (δ 67.5, 78.9) and an exocyclic methylene carbon (δ 100.4) attached to a quaternary carbon (δ 134.3). 1D-TOCSY experiments showed the presence of the spin network: CH3(δ 1. 26, d, J= 6.5 Hz)-CH(δ 3.76 dq, J= 6.5, 3.5 Hz)-CH(δ 4.05 d, J=3.5 Hz)- in the molecule. The assignment of the methyl groups, including the methoxyl and the N-bearing methyl groups, as well as the exomethylene and the carbonyl groups was substantiated by interpretation of the 1D-proton and 2D-NMR spectroscopic data of 7, including COSY, HSQC, HMBC, and NOESY experiments. The attachment of the methoxy group at C-7 was deduced by observation of the HMBC long range correlation from the signal at δ 3.32 to C-7 (δC 78.9), while the N-methyl was assigned by the observation of the long range cross-peak from the signal at δ 3.10 to C-5 (δC 67.5) and C-3 (δC 160.2). The above evidence together with the observation of the 2J and 3J HMBC correlations from the signal of the secondary methyl group (δ 1.26) to C-7 and C-5, and the cross peak from the signals of H-5 and H-7 to that of the carbonyl carbon at δ 160.2 (C-6), indicated that a threonine moiety was present in 7.
Table 1.
1H and 13C NMR Data for Compound 7
| Position | 7
|
||||
|---|---|---|---|---|---|
| Ha | Hb | Ca,c | Cb | DEPT(m) | |
| 2 | 134.3 | 133.7 | C(s) | ||
| 3 | 160.2 | 159.5 | C(s) | ||
| 5 | 4.05 d (3.5) | 3.90 d (4.1) | 67.5 | 68.2 | CH(d) |
| 6 | 160.2 | 159.5 | C(s) | ||
| 7 | 3.76 dq (6.5, 3.5) | 3.72 qd (6.5, 4.1) | 78.9 | 79.2 | CH(d) |
| 8 | 1.26 d (6.5) | 1.28 d (6.5) | 15.5 | 16.7 | CH3(q) |
| 9 | 3.32 s | 3.34 s | 56.2 | 57.2 | CH3 (q) |
| 10 | 3.10 s | 3.13 s | 35.6 | 36.6 | CH3 (q) |
| 11 | 4.88 s 5.41 s |
4.80 d (0.8) 5.60 d (0.8) |
100.4 | 102.2 | CH2 (t) |
in CD3OD;
in CDCl3;
data generated from HMBC; m: multiplicity
500 MHz and 125 MHz for CD3OD and 400 MHz and 100 MHz for CDCl3)
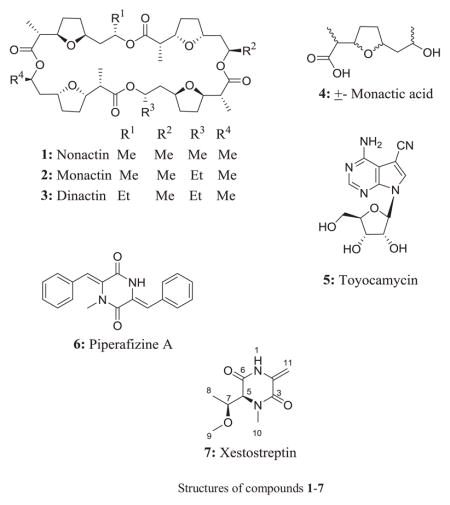
Moreover, the long range correlation from the exocyclic methylene protons to the amide carbonyl (C-3) and the four degrees of unsaturation calculated from the HRESIMS data showed that 1 was a cyclic compound. It is noteworthy that no signal was collapsed when the exocyclic methylene protons were irradiated during the 1D-NOESY experiment. The complete HMBC correlations observed in 7 allowed us to establish its structure as in Fig. 2. The relative configuration of 7 was deduced from the results obtained from 1D-NOESYspectrum (Fig. 3).
Figure 2.
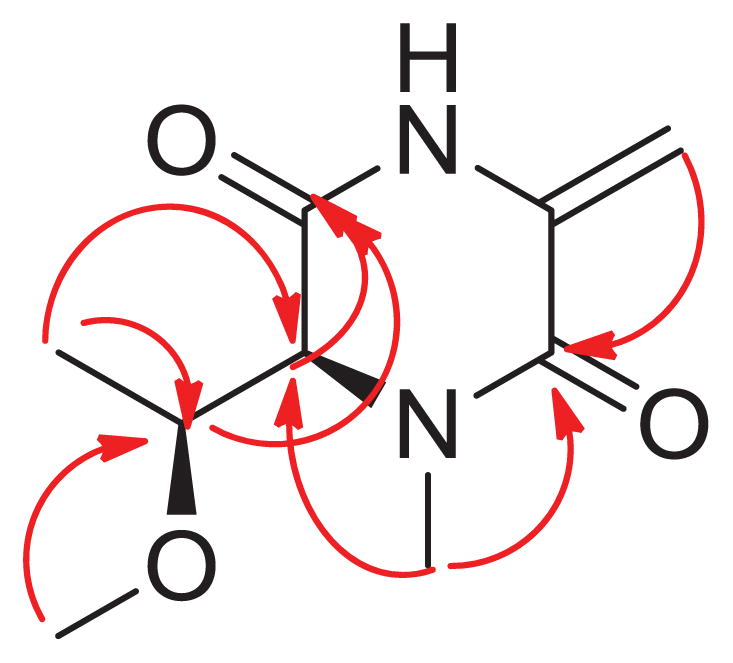
Key HMBC observed in 7
Figure 3.
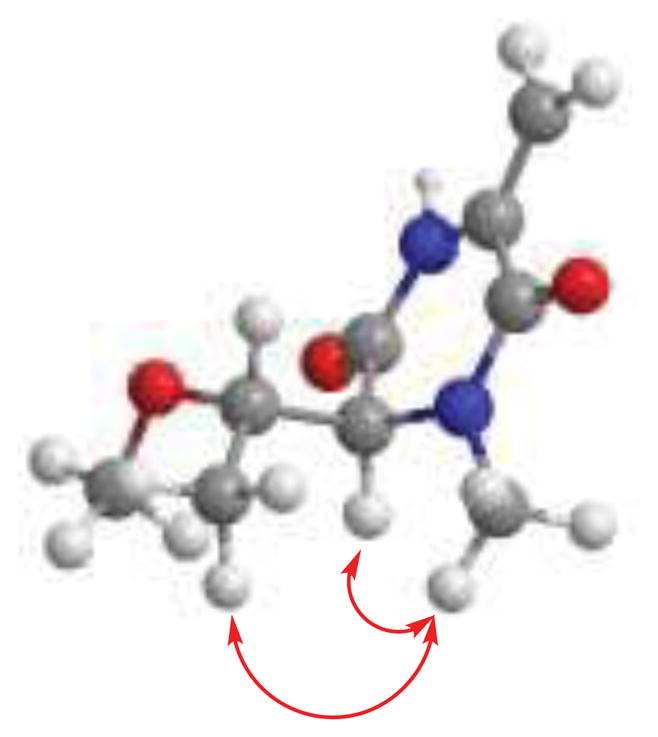
Key NOE correlations observed in 7
During our present bioguided isolation coupled with dereplication study of four selected bioactive Streptomyces strains, we identified known bioactive compounds by using prefractionation of extracts/bioassay of fractions/1H NMR/mass spectrometry analysis. The structures proposed by dereplication were confirmed by scale-up isolation. The cytotoxic compounds nonactin (1), monactin (2), dinactin (3) and toyocamycin (5) were readily identified to be responsible for the antiproliferative activity of the soil Streptomyces sp. extracts S.1 and S.2 of the present study. Compounds 1–3 isolated from S.1 inhibited the proliferation of A2780 ovarian cancer cells with IC50 values of 0.16, 0.13 and 0.26 μM, respectively. When assayed against the A2058 melanoma cell line, compounds 1–3 exhibited IC50 values of 0.26, 0.02 and 0.02 μM, respectively while IC50 values of 0.19, 0.01 and 0.01 μM, respectively were observed when tested against H522-T1 non small cancer lung cells. Our results showed that 2 and 3 exhibited some degree of selectivity in A2058 melanoma and H522-T1 cell lines. Compounds 2 and 3 were 6.5 and 13 times, respectively more active against the A2058 melanoma cell line than the A2780 cell line. This paper is an example of the use of dereplication to detect and isolate ionophores (1–4) and toyocamycin (5) which are well known cytotoxic agents produced by Streptomyces spp. Their presence in the extract can yield false positive hits during the screening. This study also demonstrated that ionophores can be easily identified by 1HNMR coupled with mass data. In addition, the phenylalanine dimer 6, previously known to be a potentiator of the cytotoxicity of vincristine, has been shown to exhibit antiplasmodial activity against the Dd2 strain of P. falciparum (IC50 6.57 μM) for the first time. Ultra-short peptides (3–4 amino acids) containing one or two phenylalanine residues have exhibited antimalarial activity11 and plasma phenylalanine has been found to be elevated in severe malaria.12 The presence of phenylalanine in 6 may contribute to the activity. More data is needed to elucidate the role of phenylalanine in the inhibition of the growth of the malaria parasite. The new compound 7, which is a methylated cyclic dipeptide from the condensation of threonine and alanine, exhibited weak antimalarial activity with an IC50 value of 50.5 μM.
In summary, this work confirms the value of using the Dictionary of Natural Products with included 1H NMR spectra and the Marinlit databases to dereplicate bioactive microbial extracts. During this study the compounds were isolated in order to confirm their structures and to demonstrate the efficacy of the method.
Supplementary Material
Acknowledgments
This project was supported by the National Center for Complementary and Alternative Medicine under award 3U01TW000313-19S1 as a supplement to Cooperative Agreement U01 TW000313-19 from the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart, Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, with the International Cooperative Biodiversity Groups. This support is gratefully acknowledged. This work was also supported by the National Science Foundation under Grant no. CHE-0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant no. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank Mr. Bill Bebout for obtaining the mass spectra. We thank The Ohio State University, College of Pharmacy NMR facility at Ohio State University for facilitating the acquisition of a part of the NMR spectra. Work in the Florida Keys was supported by grants from the National Science Foundation Microbial Observatories Program (MCB-0238515) and the Microbial Interactions and Processes Program (MCB-0703467) to RTH.
Footnotes
The authors declare no competing financial interest.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/....
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Greenwood BM, Fidock DA, Kyle DE, Kappe SHI, Alonso PL, Collins FH, Duffy PE. J Clin Invest. 2008;118:1266. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh T-NN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Abul Faiz M, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CCA, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. Nat Genet. 2015;47:226. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cragg GM, Newman DJ. Phytochem Rev. 2009;8:313. [Google Scholar]
- 4.Crevelin EJ, Crotti AEM, Zucchi TD, Melo IS, Moraes LAB. J Mass Spectrom. 2014;49:1117. doi: 10.1002/jms.3432. [DOI] [PubMed] [Google Scholar]
- 5.Forner D, Berrue F, Correa H, Duncan K, Kerr RG. Anal Chim Acta. 2013;805:70. doi: 10.1016/j.aca.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Kingston DGI. J Nat Prod. 2011;74:496. doi: 10.1021/np100550t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lang G, Mayhudin NA, Mitova MI, Sun L, van der Sar S, Blunt JW, Cole ALJ, Ellis G, Laatsch H, Munro MHG. J Nat Prod. 2008;71:1595. doi: 10.1021/np8002222. [DOI] [PubMed] [Google Scholar]
- 8.Kamei H, Oka M, Hamagishi Y, Tomita K, Konishi M, Oki T. J Antibiot. 1990;43:1018. doi: 10.7164/antibiotics.43.1018. [DOI] [PubMed] [Google Scholar]
- 9.Ogasawara M, Hasegawa M, Hamagishi Y, Kamel H, Oki T. J Antibiot. 1992;45:129. doi: 10.7164/antibiotics.45.129. [DOI] [PubMed] [Google Scholar]
- 10.Xestostreptin (7): Amorphous white powder; [α]D21 +12 (c 0.1, MeOH); IR νmax: 2924, 1723, 1686, 1621, 1461, 1286, 1128 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3), see Table 1; HRESIMS m/z 199.1068 [M+H]+ (required for C9H15N2O3+, m/z 199.1077).
- 11.Perez-Picaso L, Velasco-Bejarano B, Aguilar-Guadarrama AB, Argotte-Ramos R, Rios MY. Molecules. 2009;14:5103. doi: 10.3390/molecules14125103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopansri BK, Anstey NM, Stoddard GJ, Mwaikambo ED, Boutlis CS, Tjitra E, Maniboey H, Hobbs MR, Levesque MC, Weinberg JB, Granger DL. Infect Immun. 2006;74:3355. doi: 10.1128/IAI.02106-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.