

Abstract
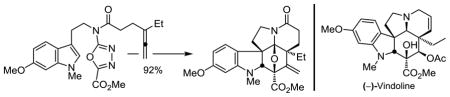
It is reported that an allene dienophile can initiate a tandem intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles, that the intermediate cross-conjugated 1,3-dipole (a carbonyl ylide) can participate in an ensuing [3 + 2] dipolar cycloaddition in a remarkably effective manner, and that the reaction can be implemented to provide the core pentacyclic ring system of vindoline. Its discovery improves a previous total synthesis of (−)-vindoline and was used in a total synthesis of (+)-4-epi-vindoline and (+)-4-epi-vinblastine that additionally enlists an alternative series of late-stage transformations.
Vinblastine (1) along with vincristine (2) are the most widely recognized members of the bisindole Vinca alkaloids as a result of their long standing clinical use as antitumor drugs (Figure 1).1–4 In addition to being among the first natural products whose structures were determined by X-ray crystallography, they were also among the first for which X-ray was used to establish their absolute configuration.5 Vindoline (3), a major alkaloid of Cantharanthus roseus, constitutes the most complex lower half of vinblastine and serves as both a biosynthetic2 and synthetic6 precursor to the natural product.
Figure 1.

Natural product structures.
Previously, we reported the development of a concise total synthesis of (−)- and ent-(+)-vindoline7–9 based on a tandem in-tramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole10–16 to assemble the pentacyclic skeleton with incorporation of all the necessary functionality and stereochemistry. The extension of this methodology to the preparation of a series of related natural products,17,18 and the subsequent development of an asymmetric total synthesis19 of vindoline followed shortly thereafter. This work along with the use of a biomimetic Fe(III)-promoted coupling of vindoline with catharanthine20 and the development of a subsequent in situ Fe(III)/NaBH4-mediated free radical alkene oxidation for C20′-alcohol introduction18,21–23 allowed for their single-step incorporation into total syntheses of vinblastine, related natural products including vincristine, and key analogues in routes as short as 8–12 steps.
These past efforts used the enol ether dienophiles 4 and 6 to initiate an oxadiazole [4 + 2]/[3 + 2] cycloaddition in which the resulting benzyl ether in the cascade cycloadducts 5 or 7 was used to install the vindoline C4-acetoxy group (Figure 2).7 While 4 permitted the direct introduction of the naturally occurring C4-OAc stereochemistry, 6 provided the C4 epimer, requiring a subsequent inversion of configuration at this center. The cyclization of 6 proceeds in excellent yield when warmed in triisopropylbenzene (TIPB), affording 7 as a single diastereomer. In contrast, the cyclization of 4 proceeds more slowly and requires more dilute reaction conditions, providing the cycloadduct 5 (53%) as a single diastereomer albeit in more modest conversions. This results from a retarded rate of the ensuing [3 + 2] cycloaddition reaction derived from a destabilizing electrostatic interaction of the benzyl ether oxygen with the oxido bridge oxygen present with 5 but not 7. Thus, although 4 directly provides the preferred cycloadduct 5 for use in the synthesis of vindoline, it proved less attractive to implement due to this less effective behavior in the cycloaddition cascade.
Figure 2.

Key step in previous work.
As a consequence of developments arising from their use in the discovery of several new classes of vinblastine analogues with properties that merit further study,15–17,22 we have continued to explore improvements in this first generation synthesis of (−)-vindoline. Herein, we report an additional and remarkably effective tandem intramolecular [4 + 2]/[3 + 2] cycloaddition reaction in which an allene dienophile initiates the cycloaddition cascade. The resulting cascade cycloadduct, which bears an exocyclic olefin, permits the expedient, improved, and divergent syntheses of not only vindoline, but also its C4 epimer. Recent exploration of substrates that bear alternative dienophiles and tethers provided the unanticipated discovery that an allene not only initiates the oxadiazole cycloaddition cascade, but its use provides the desired cycloadduct in superb yields (Scheme 1). The initiating Diels–Alder reaction between the tethered allene and the electron-deficient 1,3,4-oxadiazole affords an initial cycloadduct that undergoes loss of N2 to provide an unusual cross-conjugated 1,3-dipole (carbonyl ylide) that most might expect to be an unmanageable intermediate. The ensuing 1,3-dipolar cycloaddition reaction proceeds remarkably well and with a regioselectivity that is dictated by the linking tether, but reinforced by the intrinsic polarity of the reacting partners. Its exclusive diastereoselectivity is derived from an indole endo [3 + 2] cycloaddition, in which the dipolarophile is directed to the face opposite the newly formed six-membered ring. Four C–C bonds, three rings, five stereocenters, and the pentacyclic skeleton of vindoline are assembled in a single transformation.
Scheme 1.

Key [4 + 2]/[3 + 2] Cycloaddition Cascade
The cycloaddition cascade reaction of 8 proceeds cleanly to provide a single diastereomer 9 whose structure was first established spectroscopically and confirmed with a single crystal X-ray structure determination.24 Like the behavior of 4 and 6,7 the reaction exhibits a clear concentration dependence (Scheme 1) where an increase in the substrate concentration has the impact of lowering the yield. This is indicative of a competitive intermolecular reaction, presumably of the intermediate 1,3-dipole. When optimized (48 h), the reaction provided the single diastereomer 9 in excellent yield (92%), competitive and faster than the reaction of 6 and exceeding that of 4. Even when run for only 9 h, a 67% yield of 9 was obtained along with 30% of recovered 8 (96% yield brsm), indicating that the rate of reaction of 8 is substantially faster than either that of 4 or 6.
Because of the cross-conjugated nature of the intermediate 1,3-dipole and the range of conceivable alternative reaction pathways available, its clean and effective participation in the indole [3 + 2] dipolar cycloaddition is especially remarkable. Computational studies (AM1) on the core 1,3-dipole indicate that the olefin is in conjugation with the 1,3-dipole, making the clean cycloaddition behavior of the intermediate that much more significant (Figure 3). Although not exhaustively searched, we were unable to locate a reported carbonyl ylide that involves such an unlikely cross-conjugated structure.
Figure 3.
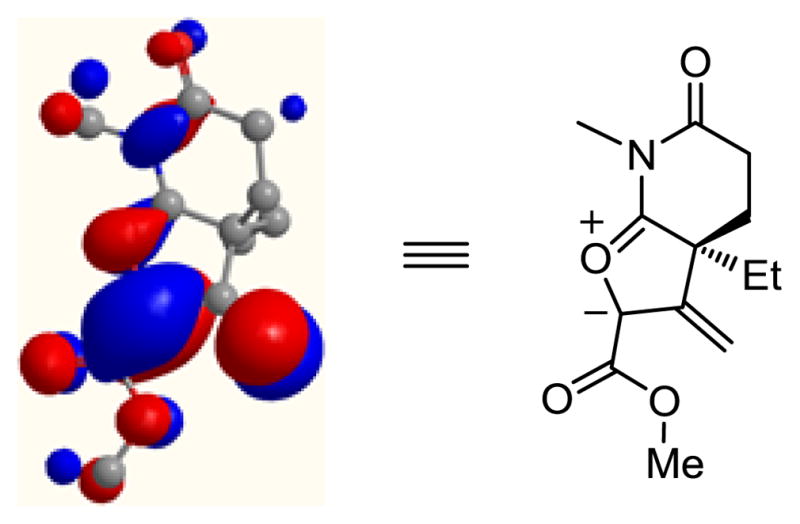
HOMO (AM1) of intermediate core 1,3-dipole.
Finally, the structure of the cascade cycloadduct 9, in our opinion, is notable. The central six-membered ring formed under the reaction conditions contains four quaternary centers, three of which are contiguous, an oxido bridge, and a sp2 hybridized carbon, making it both a strained and sterically congested oxanorbornane (see Supporting Information Figure S1).
Oxidation of the exocyclic double bond of cycloadduct 9, which is surrounded by quaternary centers, initially proved challenging. Traditional methods such as ozonolysis and the Johnson-Lemieux oxidation failed to provide the desired ketone while the classical OsO4-mediated dihydroxylation reaction conducted under Upjohn conditions (NMP/cat. OsO4) failed to yield the desired diol, most likely the result of the steric hindrance surrounding this site. However, it was found that dihydroxylation using Corey’s method (2:1 DMAP-OsO4) provided the diol remarkably effectively (Scheme 2).25 Typically, the intermediate diol 10 was not isolated and was carried directly into the subsequent oxidative cleavage reaction. Characterization of the intermediate diol 10 indicated that it is formed as a single diastereomer (81%), whose structure and stereochemistry were established in a single crystal X-ray structure determination.24 Intermediate 10 is remarkable in that it bears five contiguous quaternary centers and eleven substituents around the central six-membered ring, which is an oxanorbornane that bears four endo axial and six total α-face (endo) substituents (Figure 4). Additionally, diol 10 possesses the same C4-alcohol stereochemistry found in vindoline that arises through dihydroxylation from the sterically less hindered exo face, making it an especially attractive intermediate with which to access vindoline and vinblastine analogues containing the additional C4-hy-droxymethyl group or its derivatives.
Scheme 2.

Figure 4.

X-ray structure of 10.
Because of the surrounding steric constraints, diol 10 was not amenable to standard periodate-promoted oxidative cleavage methods (NaIO4). Rather, Pb(OAc)4 in toluene/methanol (1:1)26 was employed to provide the ketone 11 in 73% yield over two steps (Scheme 2). After chromatographic resolution of the en-antiomers of 11 (ChiralCel OD, α = 1.55, 15% i-PrOH/hexane), α-hydroxylation of the lactam enolate with (TMSO)2 followed by in situ protection of 12 (TIPSOTf/Et3N) provided silyl ether 13 (51%), which we previously converted to (−)-vindoline7 formally completing an alternative synthesis that now enlists an initiating allene dienophile in a remarkable [4 + 2]/[3 + 2] cycloaddition cascade.
With straightforward access to ketone 11, we also took the opportunity to prepare the C4 epimer of vindoline by total synthesis, exploring alternative late-stage conversions reversing the order of Δ6,7-double bond introduction, lactam carbonyl removal, and oxido bridge ring opening that could be applied to vindoline as well. Diastereoselective reduction of 11 with NaBH4 (MeOH/CH2Cl2, −78 °C, 10 min, 84%) or LiAlH(Ot-Bu)3 (THF, 0 °C, 3 h, 71%) proceeded smoothly to exclusively provide 14 (Scheme 3). Conversion of 14 to a mixture of α-selenides 15 (82%, ca. 8:1 β:α) was accomplished by treatment of the lactam enolate with diphenyl diselenide (LDA, THF, 3 h, −78 °C) without protection of the free alcohol. The mixture of selenides 15 was O-acetylated upon treatment with a 1:1 solution of Ac2O and pyridine (cat. DMAP, 16 h, 23 °C, 87%), and subsequently subjected to treatment with excess aqueous H2O2 in THF (2 h, 23 °C) to provide the α,β-unsaturated lactam 17 (92%). These latter two reactions were most conveniently conducted without the intermediate purification of the diasteromeric mixture of 16, providing 17 in further improved overall yield (85% for two steps). Treatment of 17 with Lawesson’s reagent (toluene, 1 h, 100 °C) provided thioamide 18 (77%), which was subjected to methylation with Meerwein’s salt (Me3OBF4, CH2Cl2, 1 h, 23 °C) followed by NaBH4 reduction (MeOH, 20 min, 0 °C) of the S-methyl iminium ion in the same vessel to provide the 4-epi-vindoline (19) cleanly (63%). To the best of our knowledge, not only does this represent the first total synthesis of (+)-4-epi-vindoline,15b but it was accomplished in an efficient manner, requiring only six isolated intermediates in route to 19 from the key cycloaddition cascade product.
Scheme 3.

Total synthesis of (+)-4-epi-vindoline
For comparison purposes and with 19 in hand, the C4 epimer of vinblastine was prepared in a single additional step and used to establish the effect of this single change in the natural product. Following a procedure described earlier,15b Fe(III)-promoted coupling of (+)-19 with catharanthine (20), which proceeds with complete control of the newly formed C16′ quaternary stereochemistry, and subsequent in situ Fe2(ox)3/NaBH4-mediated free radical C20′ oxidation afforded 4-epi-vinblastine (21) and its C20′ isomer 4-epi-leurosidine (2:1 β:α diastereoselectivity) (Figure 5).
Figure 5.

Total synthesis and evaluation of 4-epi-vinblastine.
In cell growth inhibition assays against a mouse leukemia (L1210) and human colon cancer (HCT116) cell line that are used to initially examine vinblastine analogues, 4-epi-vinblas-tine (21) was found to be 10-fold less potent vinblastine and essentially equipotent with 4-desacetoxyvinblastine18 that lacks the C4 acetoxy substituent altogether (Figure 5). These two comparisons indicate that both the presence and stereochemistry of the C4 acetoxy group significantly contribute to the activity of vinblastine. The origin of this substantial effect is not clear and must be subtle since this site does not interact with tubulin, rather it forms an interface with solvent in the vinblas-tine bound complex (see Supporting Information Figure S2).
Herein, we have shown that an allene dienophile effectively initiates a 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade, that the intermediate cross-conjugated carbonyl ylide participates in the ensuing [3 + 2] dipolar cycloaddition in a remarkably effective fashion, and that the reaction can be implemented to provide the pentacyclic skeleton of vindoline in excellent yield. We used this reaction to improve our total synthesis of (−)-vindoline and to provide the first total synthesis of (+)-4-epi-vindoline, enlisting an alternative series of late-stage conversions. The latter was incorporated in a single step into 4-epi-vinblastine (13-step total synthesis) whose evaluation indicates that both the presence and stereochemistry of the C4 acetoxy group significantly contribute to the activity of vinblastine. In addition to the future use of the unique diol intermediate 10, alternative alkene functionalization reactions with 9 can be envisioned to provide key additional analogues of vindoline and vinblastine and such efforts may be disclosed in due time.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056 and CA115526, DLB). T.J.B. was a NIH postdoctoral fellow (CA165303). We especially thank Professor Arnold Rheingold and James A. Golen (UCSD) for the X-ray structures of 9 and 10.
Footnotes
Full experimental details are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Noble RL, Beer CT, Cutts JH. Ann NY Acad Sci. 1958;76:882. doi: 10.1111/j.1749-6632.1958.tb54906.x. [DOI] [PubMed] [Google Scholar]; (b) Noble RL. Lloydia. 1964;27:280. [Google Scholar]
- 2.Svoboda GH, Nuess N, Gorman M. J Am Pharm Assoc Sci Ed. 1959;48:659. doi: 10.1002/jps.3030481115. [DOI] [PubMed] [Google Scholar]
- 3.Neuss N, Neuss MN. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego, CA: 1990. pp. 229–240. [Google Scholar]
- 4.Pearce HL. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego, CA: 1990. pp. 145–204. [Google Scholar]
- 5.Moncrief JW, Lipscomb WN. J Am Chem Soc. 1965;87:4963. doi: 10.1021/ja00949a056. [DOI] [PubMed] [Google Scholar]
- 6.Kuehne ME, Marko I. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego, CA: 1990. pp. 77–132. [Google Scholar]
- 7.(a) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Z, Ishikawa H, Boger DL. Org Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Additional and prior total syntheses of vindoline, racemic total syntheses: Ando M, Büchi G, Ohnuma T. J Am Chem Soc. 1975;97:6880.Kutney JP, Bunzli-Trepp U, Chan KK, De Souza JP, Fujise Y, Honda T, Katsube J, Klein FK, Leutwiler A, Morehead S, Rohr M, Worth BR. J Am Chem Soc. 1978;100:4220.Andriamialisoa RZ, Langlois N, Langlois Y. J Org Chem. 1985;50:961. doi: 10.1021/jo01336a006.Ban Y, Sekine Y, Oishi T. Tetrahedron Lett. 1978;2:151.Takano S, Shishido K, Sato M, Yuta K, Ogasawara K. J Chem Soc, Chem Commun. 1978:943.Takano S, Shishido K, Matsuzaka J, Sato M, Ogasawara K. Heterocycles. 1979;13:307.Danieli B, Lesma G, Palmisano G, Riva R. J Chem Soc, Chem Commun. 1984:909.Danieli B, Lesma G, Palmisano G, Riva R. J Chem Soc, Perkin Trans. 1987;1:155.Zhou S, Bommeziijn S, Murphy JA. Org Lett. 2002;4:443. doi: 10.1021/ol0171618.En-antioselective total syntheses: Feldman PL, Rapoport H. J Am Chem Soc. 1987;109:1603.Kuehne ME, Podhorez DE, Mulamba T, Bornmann WG. J Org Chem. 1987;52:347.Cardwell K, Hewitt B, Ladlow M, Magnus P. J Am Chem Soc. 1988;110:2242.Kobayashi S, Ueda T, Fukuyama T. Synlett. 2000:883.
- 10.Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew Chem, Int Ed. 2006;45:620. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa H, Boger DL. Heterocycles. 2007;72:95. [Google Scholar]
- 12.Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan Z, Boger DL. J Am Chem Soc. 2006;128:10589. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DB, Miller MM, Pollack S, Boger DL. J Am Chem Soc. 2002;124:11292. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]
- 14.(a) Boger DL. Tetrahedron. 1983;39:2869. [Google Scholar]; (b) Boger DL. Chem Rev. 1986;86:781. [Google Scholar]
- 15.(a) Va P, Campbell EL, Robertson WM, Boger DL. J Am Chem Soc. 2010;132:8489. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. J Med Chem. 2013;56:483. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Tam A, Gotoh H, Robertson WM, Boger DL. Bioorg Med Chem Lett. 2010;20:6408. doi: 10.1016/j.bmcl.2010.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gotoh H, Duncan KK, Robertson WM, Boger DL. ACS Med Chem Lett. 2011;2:948. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Review: Sears JE, Boger DL. Acc Chem Res. 2015;48:653. doi: 10.1021/ar500400w.
- 18.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Sasaki Y, Kato D, Boger DL. J Am Chem Soc. 2010;132:13533. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kato D, Sasaki Y, Boger DL. J Am Chem Soc. 2010;132:3685. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Vukovic J, Goodbody AE, Kutney JP, Misawa M. Tetrahedron. 1988;44:325. [Google Scholar]; (b) Sundberg RJ, Hong J, Smith SQ, Sabato M, Tabakovic I. Tetrahedron. 1998;54:6259. [Google Scholar]; (c) Gotoh H, Sears JE, Eschenmoser A, Boger DL. J Am Chem Soc. 2012;134:13240. doi: 10.1021/ja306229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leggans EK, Barker TJ, Duncan KK, Boger DL. Org Lett. 2012;14:1428. doi: 10.1021/ol300173v.For additional applications, see: Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716.Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. J Med Chem. 2013;56:628. doi: 10.1021/jm3015684.Barker TJ, Duncan KK, Otrubova K, Boger DL. ACS Med Chem Lett. 2013;4:985. doi: 10.1021/ml400281w.
- 23.(a) Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J Am Chem Soc. 2014;136:1300. doi: 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC, Yabe Y, Baran PS. J Am Chem Soc. 2014;136:1304. doi: 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The structure and stereochemistry of 9 (CCDC 1412216) and 10 (CCDC 1412217) were established with racemic material by X-ray.
- 25.He F, Bo Y, Altom JD, Corey EJ. J Am Chem Soc. 1999;121:6771.Corey EJ, Sarshar S, Azimioara MD, New-bold RC, Noe MC. J Am Chem Soc. 1996;118:7851.Review: Schmidt A-KC, Stark CBW. Synthesis. 2014;46:3283.
- 26.Corey EJ, Hong B. J Am Chem Soc. 1994;116:3149. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.