

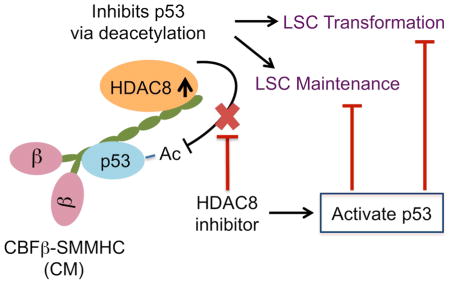
Summary
Acute myeloid leukemia (AML) is driven and sustained by leukemia stem cells (LSCs) with unlimited self-renewal capacity and resistance to chemotherapy. Mutation in the TP53 tumor suppressor is relatively rare in de novo AML; however, p53 can be regulated through post-translational mechanisms. Here, we show that p53 activity is inhibited in inv(16)+ AML LSCs via interactions with the CBFβ-SMMHC (CM) fusion protein and histone deacetylase 8 (HDAC8). HDAC8 aberrantly deacetylates p53 and promotes LSC transformation and maintenance. HDAC8 deficiency or inhibition using HDAC8-selective inhibitors (HDAC8i) effectively restores p53 acetylation and activity. Importantly, HDAC8 inhibition induces apoptosis in inv(16)+ AML CD34+ cells while sparing the normal hematopoietic stem cells. Furthermore, in vivo HDAC8i administration profoundly diminishes AML propagation and abrogates leukemia-initiating capacity of both murine and patient-derived LSCs. This study elucidates a HDAC8-mediated p53-inactivating mechanism promoting LSC activity, and highlights HDAC8 inhibition as a promising approach to selectively target inv(16)+ LSCs.
Graphical Abstract
Introduction
Acute myeloid leukemia (AML) is an aggressive bone marrow malignancy with over 20,000 new cases and 10,000 deaths each year in the United States. AML arises from leukemia stem cell (LSC) transformation as a consequence of multiple cooperative mutations or epigenetic alterations. Recurrent chromosomal abnormalities in AML frequently result in transcription factor fusion proteins that contribute to the unique etiology and prognosis of distinct cytogenetic subsets (Look, 1997). The core-binding factor (CBF) complex, consists of a DNA-binding RUNX protein and a non-DNA binding CBFβ, is a master transcriptional regulator of hematopoiesis and a frequent target of leukemia associated mutations (Speck and Gilliland, 2002) One of the common recurrent cytogenetic aberrations found in approximately 5–12% of AML patients is chromosome 16 inversion inv(16)(p13.1q22) or translocation t(16;16)(p13.1;q22) [henceforth inv(16)] (Liu et al., 1996). Inv(16) results in fusion of CBFB with the MYH11 gene, which encodes a smooth muscle myosin heavy chain (SMMHC) protein (Liu et al., 1993). The resulting fusion protein CBFβ-SMMHC (CM) retains the RUNX1 binding interface of CBFβ and the coiled-coil rod region of SMMHC. Heterozygote Cbfb-MYH11 knock-in (KI) at the Cbfb locus led to lethal defects in definitive hematopoiesis at E12.5 (Castilla et al., 1996), replicating the phenotypes of Runx1- or Cbfb-null mice (Okuda et al., 1996; Wang et al., 1996). These results suggested that CM is a dominant inhibitor of CBF during hematopoietic ontogeny. Our previous studies in a conditional CM KI mouse model further revealed that CM expression in HSCs leads to impaired differentiation of multiple hematopoietic lineages and produces pre-leukemic stem and progenitors that are at risk for LSC transformation (Castilla et al., 1996; Kuo et al., 2006; Kuo et al., 2008; Zhao et al., 2007). However, the molecular mechanism underlying the leukemogenic function of CM has been elusive.
AML is maintained and propagated by LSCs that are relatively resistant to chemotherapy and can persist as potential sources of relapse. Although patients with inv(16) AML have a relatively favorable prognosis, only approximately half of them eventually achieve long-term survival with the standard chemotherapy regimens (Prébet et al., 2009; Ustun and Marcucci, 2015). Molecular insights into leukemogenic mechanisms are necessary to design new LSC-targeted therapies to reduce relapse and improve outcomes. TP53 mutations are relatively rare in de novo AML (approximately 10%); however, TP53 mutation is associated with complex karyotypes, drug resistance and dismal outcome (Rücker et al., 2012; Haferlach et al., 2008). Loss of p53 has also been shown to promote AML pathogenesis in mice by enabling aberrant self-renewal (Zhao et al., 2010). The functions of p53 are coordinately modulated by a number of post-translational modifications including acetylation (Dai and Gu, 2010). Given the low TP53 mutation rate, alternative mechanisms affecting p53 protein stability or post-translational modification are possibly involved in disrupting p53 function during AML pathogenesis.
Histone deacetylases (HDACs) are a family of enzymes that catalyze the removal of acetyl moieties from lysine residues in a variety of histones proteins and transcription factors including p53. HDAC8 is a class I HDAC that is overexpressed in multiple tumor types, including neuroblastoma, glioma (Oehme et al., 2009) and childhood acute lymphoblastic leukemia (Moreno et al., 2010). Although HDAC8 has been shown to interact with the CM chimeric protein as part of a transcriptional repressor complex (Durst et al., 2003), its functional role in AML pathogenesis is unclear. In this study, we uncovered a HDAC8-mediated post-translational p53-inactivating mechanism underlying CM-associated LSC transformation and maintenance. We investigated the functional contribution of HDAC8 in human AML stem/progenitor cell survival and propagation, and evaluated the efficacy of HDAC8-selective inhibitors in targeting murine and human AML LSCs in vivo.
Results
CM fusion protein binds to p53 and impairs p53 acetylation
The activity of p53 is normally tightly controlled and is rapidly induced upon genotoxic stress triggered by radiation or chemotherapy. Acetylation of the p53 protein, critical for its stability and transcriptional activity, is rapidly induced upon p53 activation (Dai and Gu, 2010). We have previously generated a conditional CM KI mouse model (Cbfb56M/+/Mx1-Cre), which expresses CM under the endogenous Cbfb promoter after polyinosinic polycytidylic acid (pIpC) treatment (Kuo et al., 2006). Western blot analysis using an antibody against an acetylated (Ac)-form of p53 (K379) revealed that Ac-p53 levels were largely reduced in CM pre-leukemic (2 weeks after pIpC) bone marrow (BM) cells treated with γ-irradiation (IR, 3Gy) compared to similarly treated control BM (Figure 1A). Time course analysis revealed that the initial acetylation of p53 occurred (2 h), however, p53 was rapidly deacetylated in the presence of CM (Figure 1A). To verify whether this is directly related to CM expression, we transduced a myeloid progenitor cell line 32D (p53 intact) with MSCV-ires-GFP (MIG) expressing FLAG-tagged CM (32D-CM) or normal CBFβ (32D-CBFβ). The levels of Ac-p53 were consistently reduced in 32D-CM compared to 32D-CBFβ cells (Figure 1B). In addition, expression of CM by transducing Cbfb56M/+ BM cells with a MIG-Cre vector readily reduced Ac-p53 induction (Figure 1C), suggesting this likely to be a direct effect of CM. Furthermore, knocking-down CM using small-hairpin (sh)-RNAs against the Cbfb-MYH11 sequence rapidly restored Ac-p53 induction in 32D-CM cells (Figure 1D). Similarly, silencing CM in mouse AML cells significantly induced p53 target gene expression (Figure 1E, S1A). The transcription of p53 was not affected as CM expression in 32D cells or in primary myeloid progenitors did not cause significant changes in p53 mRNA levels (Figure S1B).
Figure 1. CM fusion protein binds to p53 and impairs p53 acetylation.

A. Western blot time course analysis of Ac-p53, p53, CM, CBFβ, β-actin after IR (3 Gy) in CM-expressing or control BM cells.
B. Western blot time course analysis of Ac-p53, p53, CM, CBFβ, HDAC8 and β-actin after IR (3Gy) in 32D-CM or 32D-CBFβ cells.
C. Western blot of Ac-p53, p53, CM, CBFβ, β-actin in Cbfb56M/+ BM progenitor cells transduced with MIG-Cre and stimulated with IR (3Gy, 6 h).
D. Western blot of Ac-p53, p53, CM, CBFβ, β-actin in 32D-CM cells expressing control (Ctrl)-shRNA or CM-shRNA (A3, D4) 6 h after IR.
E. Western blot of CM and β-actin (top) and relative expression of p53 target genes in sorted AML cells transduced with ctrl-shRNA or CM-shRNA (A3, D4) and induced with IR (bottom). Shown are fold change (mean ± SD) relative to ctrl-shRNA-expressing cells, performed in triplicate. *P < 0.05; **P < 0.01; ***P < 0.001.
F. Co-IP and immunoblot (IB) analysis in 32D-CM (top) or 32D-CBFβ (bottom) cells using anti-p53 or anti-mouse IgG for IP, and anti-p53 (left) or anti-CBFβ (right) for IB.
G. Representative images of Duolink in situ PLA using mouse anti-CBFβ, rabbit anti-p53 antibodies and PLA probes. Red foci indicate CM-p53 interactions (left), DAPI-stained nuclei are in blue (center) and GFP+ indicates transduced cells (right); scale bar, 10 μm.
H. Co-IP (anti-CBFβ) and IB (anti-p53 or anti-CBFβ) analysis in control BM or CM BM cells with or without IR (3Gy).
I. Co-IP (anti-p53 or IgG) and IB (anti- CBFβ or anti-p53) in inv(16)+ AML (163, 987) or non-inv(16) AML (467, 865) CD34+ cells 3h after IR.
See also Figure S1.
To determine how CM fusion protein impairs p53 activity, we examined whether CM interacts with the p53 protein. First, we performed co-immunoprecipitation (co-IP) followed by western blot analysis in 32D-CM and 32D-CBFβ cells. We found that the CM fusion protein, but not CBFβ, was pulled down together with p53 (Figure 1F, S1D). As an alternative approach, we used a proximity ligation assay (PLA) to assess intermolecular interaction in situ. Punctate red fluorescent foci were observed in 32D-CM but not 32D-CBFβ cells (Figure 1G), indicating the presence of specific interaction between CM and p53 proteins. In contrast, in situ PLA using an Ac-p53 (K379) specific antibody showed very few interacting foci (Figure S1C), suggesting that the p53 protein interacting with CM is mostly deacetylated. Nuclear and cytoplasmic fractionation revealed that CM-p53 complex is detected mainly in the nucleus (Figure S1E). Similar CM-p53 interaction was seen when RUNX1 is knocked down (Figure S1F), suggesting that this binding dose not require RUNX1. The CM-p53 complex can also be detected in primary CM pre-leukemic BM cells with or without IR (Figure 1H). Furthermore, co-IP in primary AML CD34+ cells isolated from patients showed that CM-p53 protein complex was seen specifically in inv(16)+ AML CD34+ cells and not in non-inv(16) cells (Figure 1I). Altogether, these results indicate that CM fusion protein binds to p53 and impairs acetylation and activation of p53.
HDAC8 mediates CM-induced deacetylation of p53
Consistent with previous report (Durst et al., 2003), we found that CM interacts specifically with HDAC8 and not other class I HDACs including HDAC1, HDAC2 or HDAC3 (Figure S2A, B). We next assessed whether CM forms a multimeric protein complex with HDAC8 and p53 by sequential IP followed by western blot analysis. Indeed, we were able to pull down CM, p53, and HDAC8 in a protein complex using two different antibody sequential combinations (Figure 2A). To further examine whether the CM-p53 interaction is dependent on HDAC8 binding, we generated a set of CM deletion mutants (Figure 2B, left). The CM-ΔC95 mutant lacking 95 amino acids at the C-terminus did not bind HDAC8 (Figure S2B), as previously reported (Durst et al., 2003). Co-IP and in situ PLA revealed that CM-ΔC95 can bind p53 while CM variants d134 (residues 134-236 deleted) and d179 (residues 179-221 deleted) cannot (Figure 2B, C). In addition, co-IP and in situ PLA showed that CM-p53 binding was unaffected in Hdac8 knocked-down 32D-CM cells (Figure 2C, D, S2C). These results suggest that CM-p53 interaction does not require HDAC8 binding. However, aberrant p53 deacetylation associated with CM depends critically on Hdac8 because knocking-down Hdac8 led to a robust increase in Ac-p53 induced by IR (Figure 2E). In addition, CM deletion mutants ΔC95, d134 or d179 that were unable to bind either HDAC8 or p53 had no effect on Ac-p53 induction compared to control (FLAG or CBFβ) 32D cells (Figure 2F, S2D). Quantitative (q) RT-PCR analysis show that induction of p53 target genes including p21 Cdkn1a, Mdm2, Bid, Bax, and Gadd45b were restored in CM-ΔC95 cells compared to full-length CM-expressing cells (Figure 2G). Collectively, these results indicate that although binding to p53 and HDAC8 occurs through distinct regions of the CM protein, simultaneous interaction with HDAC8 and p53 is required for aberrant deacetylation and inactivation of p53.
Figure 2. CM fusion protein recruits HDAC8 and p53 in a protein complex and promotes the deacetylation of p53 by HDAC8.

A. Sequential co-IP (1° IP anti-HDAC8 or anti-p53, 2° IP anti-CBFβ) and IB (anti-p53 or anti-HDAC8) analysis in 32D-CBFβ or CM cells that were not IR (left) or IR (3 Gy)-treated (right).
B. Illustration of CM deletion variants (left) used in the co-IP (anti-p53) and IB (anti-CBFβ or anti-p53) analysis (right). Red arrows indicate the expected size of CM variants.
C. Representative images of in situ PLA in 32D FL-CM, d134, d179, ΔC95, Ctrl- or Hdac8-shRNA (sh1 or sh2) expressing cells using mouse anti-CBFβ, rabbit anti-p53 and PLA probes. CM-p53 interactions are shown in red (top), DAPI staining is in blue (center) and the GFP reporter indicates transduced cells (bottom); scale bar, 10 μm.
D. Co-IP (IgG or anti-p53) and IB (anti-CBFβ or anti-p53) in 32D-CM cells expressing Ctrl- or Hdac8-shRNA (sh1 or sh2).
E. Western blotting of Hdac8, Ac-p53 (K379), p53 and β-actin in 32D-CM cells expressing Ctrl- or Hdac8-shRNA after IR (3Gy, 6h).
F. Western blotting of Ac-p53, p53 in CM-, ΔC95-, CBFβ- or FLAG-expressing 32D cells before or after IR (3Gy).
G. Fold induction of p53 target genes in CBFβ, CM or CM-ΔC95 expressing cells, 24h after 3Gy IR. Bars represent mean ± SD duplicated assays and two experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
See also Figure S2.
HDAC8 promotes CM-induced LSC transformation and cell survival through deacetylase activity
To determine the contribution of HDAC8 in CM-induced AML LSC transformation, we generated a floxed Hdac8 (Hdac8f) allele in which exon 3 was flanked by loxP sites (Figure S3A). Mice carrying Hdac8f/f were crossed with Cbfb56M/+/Mx1-Cre (C56Bl6 strain) and induced with pIpC as previously described (Kuo et al., 2006). Successful induction of Hdac8 deletion in Hdac8KO/Cbfb56M/+/Mx1-Cre mice was confirmed by PCR (not shown) and western blot analysis (Figure 3A). CM/Hdac8KO pre-leukemic BM progenitors show significantly higher levels of Ac-p53 upon IR (Figure 3B, S3B), consistent with HDAC8 mediating CM-associated p53 deacetylation. Similar to previous studies, C56BL/6 CM KI mice spontaneously develop AML with 100% penetrance and a median survival of 122 days. Notably, deletion of Hdac8 (CM/Hdac8KO) significantly (p<0.0001) reduced AML incidence (13.6%) and prolonged disease-free survival (Figure 3C). These results indicate that HDAC8 promotes LSC transformation and AML progression.
Figure 3. Hdac8 deletion diminishes CM-induced LSC transformation and promotes p53 activation.

A. Western blot analysis of Hdac8, CM, CBFβ in BM cells from Cbfb56M/+/Mx1-Cre (CM) or CM/Hdac8KO mice 2 weeks after 7 doses of pIpC treatment.
B. Western blotting of Ac-p53, p53 before or after (2h) IR (3Gy) in pre-leukemic CM or CM/Hdac8KO BM cells.
C. Kaplan-Meier survival curve of induced CM (n=23; median survival 122 days) or CM/Hdac8KO mice (n=56) mice monitored up to 1 year.
D. Western blotting of Ac-p53, p53, Hdac8 and β-actin in 32D-CM cells treated with indicated dose of HDAC8i PCI-34051 (μM), 22d (μM) or Nutlin-3 (2.5 μM) for 6 h.
E. Western blotting of Ac-p53, p53, Ac-Smc3, Smc3, and β-actin in 32D-CM cells treated with 22d (5 μM), MS-275 (2.5 μM), TV6 (2.5 μM), PCI-24781 (200 nM) or Nutlin-3 (2.5 μM).
F. Fold activation of p53 targets in 32D-CM cells treated with HDAC8i PCI-34051 (10 μM) or 22d (10 μM) for 16 h, relative to levels in vehicle-treated cells (dashed line). Shown are mean ± SD of triplicated qRT-PCR assays and two experiments.
G. Fold activation of p53 targets in primary CM (n=4) or CM/Hdac8KO (n=5) progenitors cells treated with the HDAC8i 22d (10 μM) for 16 h, relative to levels in vehicle treated cells (dashed line). Mean ± SD of triplicated qRT-PCR assays are shown.
H. Survival inhibition dose-response curve and IC50 of HDAC8i 22d for 32D-CBFβ, 32D-CM or 32D-CM cells overexpressing HDAC8.
I. Western blot analysis of Ac-p53, p53, Ac-SMC3, SMC3, HDAC8 and β-actin in 32D-CM cells with or without HDAC8 overexpression (O/E) treated with 22d (0, 2.5, 5, 10 μM).
J. Fold induction of luciferase reporter under the control of p53 responsive elements in K562 cells co-transfected with CM and p53-WT or p53-8KR mutant, and treated with 22d (10 μM). Dashed line indicates levels in vehicle treated cells. Mean ± SD of four assays in two experiments are shown.
*P < 0.05; **P < 0.01; ***P < 0.001; see also Figure S3.
To test whether p53 acetylation and activity could be modulated by HDAC8 deacetylase activity, we used HDAC8-selective pharmacological inhibitors (HDAC8i) including PCI-34051 (Balasubramanian et al., 2008) and compound 22d (Huang et al., 2012) designed to block HDAC8 catalytic activity. Treatment with both HDAC8i compounds remarkably enhanced Ac-p53 in 32D-CM cells (Figure 3D, lanes 3, 8, 9 from left). Because p53 protein levels were also increased upon HDAC8i treatment, we included the Mdm2 inhibitor Nutlin-3 to stabilize the p53 protein. Treatment of HDAC8i (PCI-34051 or 22d) in combination with Nutlin-3 elevated Ac-p53 compared to Nutlin-3 alone (Figure 3D, lanes 4–6; 10–12 from left), suggesting that the enhanced Ac-p53 by HDAC8i is not a result of p53 protein stabilization. In fact, 22d differentially increased Ac-p53 as well as the HDAC8-specific substrate Ac-SMC3 (Deardorff et al., 2012) compared to other class I HDAC (MS-275), class III HDAC (TV6) or broad-spectrum (PCI-24781) inhibitors with limited HDAC8 inhibitory activity (Figure 3E, S3C). In addition, HDAC8i (PCI-34051 or 22d) treatment significantly induced expression of multiple p53 targets as determined by qRT-PCR analysis (Figure 3F). This is indeed p53-dependent since knocking down p53 significantly reduced the effect (Figure S3D, E). Similarly, Hdac8 deletion significantly decreased p53 targets induction by 22d (Figure 3G). Activation of p53 upon 22d treatment was accompanied by a dose-dependent increase in apoptosis of 32D-CM cells (IC50=4.25 μM) compared to 32D-CBFβ cells (IC50=7.54 μM). This enhanced apoptosis and Ac-p53 can be rescued by overexpression of HDAC8 (IC50=9.15 μM; Figure 3H–I). In addition, primary CM/Hdac8KO BM cells (IC50=7.36 μM) were significantly more resistant to 22d-induced apoptosis compared to CM BM cells (IC50=4.99 μM; Figure S3F). To further substantiate that this is mediated by p53 activation, we expressed CM in BM cells from p53ERTAM KI mice carrying a 4-hydroytamoxifen (4-OHT) inducible form of the p53 protein (Christophorou et al., 2005). Restoring p53 activity upon 4-OHT treatment significantly increased apoptosis and the sensitivity to 22d (Figure S3G). Furthermore, human K562 cells (p53 inactive) were significantly (p=0.0024) more sensitive to 22d when co-expressing CM and wild type (WT) p53 (IC50=9.48 μM) compared to an acetylation sites-mutated (8KR; IC50=21.03 μM) form of p53 (Figure S3H, I). Using a p53-luciferase reporter construct under the control of p53 responsive elements (Dai et al., 2004), we show that p53 transcription activity is significantly induced by 22d in K562 cells rescued with p53-WT but not p53-8KR (Figure 3J). Altogether, these results provide genetic evidence that 22d induces p53 activation and apoptosis through inhibition of HDAC8.
Inhibition of HDAC8 selectively induces apoptosis of human inv(16)+ AML stem and progenitor cells
To evaluate the role of HDAC8 in human AML, we first examined the expression of HDAC8 in primary human CD34+ cells. The levels of HDAC8 mRNA were signiifcantly higher in inv(16)+ AML CD34+ cells (n=7; p=0.0003) compared to normal (NL) mobilized CD34+ peripheral blood stem cells (PBSCs; n=7) or non-inv(16) AML (n=19) CD34+ cells (Figure 4A). Treatment with HDAC8i (22d, 48h) significantly reduced the proliferation of inv(16)+ CD34+ cells relative to normal CD34+ cells (Figure S4A, inv(16)+ AML n=9; normal n=7). More importantly, 22d selectively induced apoptosis of inv(16)+ AML CD34+ cells (IC50=10.65 μM) compared to t(8;21)+ CBF AML (IC50=25.99 μM; p=0.0176), other non-inv(16) AML CD34+ (IC50=45.56 μM; p=0.0011) or normal CD34+ (IC50=99.92 μM; p<0.0001) cells (Figure 4B, C). Meanwhile, treatment with 22d (10 μM) did not affect the short- or long-term engraftment capacity of normal CD34+ cells (Figure S4B and not shown).
Figure 4. Inhibition of HDAC8 selectively induces p53-dependent apoptosis in inv(16)+ AML stem/progenitor cells.

A. Relative expression of HDAC8 in normal (NL) PBSC (n=7; mean indicated by dashed line), inv(16)+ AML (n=7) or non-inv(16)+ AML (n=19) CD34+ cells. Each dot represents the average of triplicate qRT-PCR assays from an individual patient and line indicates the mean ± SEM of all samples.
B. Percent survival of NL (n=7), inv(16)+ AML (n=12), t(8;21) AML (n=3) or other non-inv(16) AML (n=4) CD34+ cells treated with HDAC8i 22d for 48 h, measured by Annexin V labeling. Each dot represents an individual subject and lines indicate the mean ± SEM.
C. Survival inhibition dose-response curve and IC50 of HDAC8i 22d for NL, inv(16)+ AML, t(8;21) AML or other non-inv(16) AML CD34+ cells as described in B.
D. Western blots of Ac-p53 (K382), p53 and β-actin in inv(16)+ AML CD34+ cells treated with 22d (10 μM) or Nutlin-3 (2.5 μM) for 6 h. Shown are representative results from four patients.
E. Fold induction of p53 target genes in inv(16)+ AML CD34+ (n=9–13) or NL CD34+ (n=7) cells treated with 22d (10 μM) for 16 h, relative to the levels in vehicle-treated cells (dashed line). Each dot represents the mean of triplicated qRT-PCR assay for an individual subject and the lines indicate the mean ± SEM.
F. Representative FACS plot of Annexin V/DAPI labeling in inv(16)+ AML CD34+ cells expressing control (ctrl) or p53 shRNA (GFP+) and treated with 22d for 48 h.
G. Relative survival of sorted GFP+ inv(16)+ AML CD34+ cells expressing ctrl- or p53-shRNA treated with 22d for 48 h (top). Each dot represents an individual patient and the lines indicate mean ± SEM. Dose-response curve and IC50 of shctrl- (solid line) or shp53-cells (dashed line) treated with 22d (bottom).
H. Percent survival of inv(16)+ AML (n=3–7) CD34+ cells treated with 22d (5 or 10 μM) or combined with DNR (50 or 100 nM) or Ara-C (0.2, 1, 5μM).
*P < 0.05; **P < 0.01; ***P < 0.001; ns: not significant. See also Figure S4 and Table S1.
Next, we assessed whether HDAC8i could selectively activate p53 in inv(16)+ AML CD34+ cells. Indeed, enhanced Ac-p53 (K382) upon 22d (6h) treatment were consistently observed in all inv(16)+ patient samples but not in non-inv(16) AML (p53 non-mutated) or normal CD34+ cells (Figure 4D, S4C and not shown). Expression of p53 target genes, particularly apoptosis-related genes, were also significantly induced in inv(16)+ CD34+ cells (n=9–13) relative to normal (NL; n=7) CD34+ cells (Figure 4E). The apoptosis-inducing effect of 22d was significantly reduced in p53 knocked-down (Figure S4D) inv(16)+ CD34+ cells (IC50=54.6 μM; p=0.0079) compared to non-silenced control (IC50=16.62 μM) (Figure 4F–G; n=5), suggesting that p53 is critical for the apoptotic effects of 22d. Furthermore, treatment with 22d (5 or 10 μM) significantly enhanced the sensitivity to cytarabine (Ara-C) or daunorubicin (DNR), commonly used chemotherapeutics for AML treatment (Figure 4H). These results suggest that inv(16)+ AML stem and prognitors are selectively dependent on HDAC8 activity for survival, and that inhibiting HDAC8 activates p53 thereby sensitizing for apoptosis.
Pharmacologic inhibition of HDAC8 abrogates AML propagation and eliminates LSC leukemia-initiating capacity
AML LSCs are functionally defined by their capacity to engraft and reproduce AML disease in serial transplants. To test whether HDAC8 inhibition could affect AML engraftment and LSC leukemia-initiating capacity, we used a CM AML mouse model with a tdTomato+ Cre-reporter (Cbfb56M/+/Mx1-Cre/tdTomato+). These mice develop transplantable AML 3–6 months after pIpC induction and allow easy tracking of CM-expressing AML cells (dTomato+/cKit+). First, we examined the effect of 22d treatment on AML cell engraftment capacity after ex vivo exposure (Figure 5A). AML engraftment was clearly inhibited by 22d treatment, as evidenced by low amounts of AML cells detected in the peripheral blood (PB), spleen and BM of mice receiving 22d-treated cells (Figure 5B–F, S5A–B). Over 80% (9 of 11) of vehicle-treated recipients succumbed to lethal AML (median survival 68 days) while no AML reoccurrence was observed in 22d-treated group (n=8) monitored up to 8 months (Figure 5G; p=0.0007). In addition, we assessed changes in the engraftment capacity of AML cells by transplanting equal numbers of cells (2×106) that survived the 22d or vehicle treatment (48h). Similarly, engraftment levels were significantly lower in recipients of 22d-treated cells compared to the vehicle control group in which 2 mice died of aggressive AML before analysis (Figure 5H–I, S5C–D). None of the 22d-treated recipients developed leukemia compared to lethal AML seen in 100% (n=4) vehicle control mice (Figure 5J; p=0.0091). These results indicate that HDAC8i treatment diminished AML engraftment capacity on a per cell basis.
Figure 5. Ex vivo treatment of HDAC8i 22d diminishes AML engraftment and leukemia initiation.

A. Schematic illustration of the experimental design. AML cells were isolated from pIpC-induced Cbfb+/56MMx1Cre/tdTomato+ moribund mice, treated with 22d or vehicle for 48 or 72 h and transplanted into congenic mice. Engraftment of AML in PB was monitored over time (4, 8 weeks). Mice were analyzed for BM and spleen engraftment at 8 weeks or monitored for leukemia onset and survival up to 8 months.
B. Engraftment of AML cells in the PB at 4 (n=7; P=0.0006) or 8 weeks (vehicle, n=5; 22d, n=7; P=0.0025). Shown are mean ± SEM.
C. Representative images of spleens from recipients of vehicle-treated (top) or 22d-treated cells (bottom) at 8 weeks.
D. Representative FACS plots and frequencies of engrafted dTomato+/cKit+ AML cells in the BM at 8 weeks.
E. Frequency of AML cells in the BM of recipients of vehicle treated (n=5) or 22d treated cells (n=7). Each dot represent results from an individual mouse and lines indicate mean ± SEM. **P=0.0025
F. Frequency of AML cells in the spleen of recipients of vehicle-treated (n=5) or 22d-treated cells (n=7). **P=0.0025
G. Survival curve of mice transplanted with AML cells treated with 22d (n=8) or vehicle (n=11; ***P=0.0007).
H. The frequency of dTomato+ cells in the PB 8 weeks after transplantation of 2×106 live AML cells treated with 22d or vehicle ex vivo for 48 h (n=4). Each dot represents results from individual mice and line indicate mean ± SEM. *P=0.0357
I. The frequency of dTomato+/ckit+ cells in the BM 28 weeks after transplantation of 2×106 live cells treated with 22d (n=4) or vehicle (n=2; another 2 had died of AML prior to analysis) ex vivo for 48 h (n=4). Each dot represents results from individual mice and line indicate mean ± SEM. *P=0.0206
J. Survival curve of mice transplanted with 2×106 live AML cells treated with 22d or vehicle (n=4; **P=0.0091).
See also Figure S5.
Next, we evaluated the efficacy of in vivo administration of HDAC8i 22d. To generate cohorts of AML-bearing mice, AML cells were directly transplanted into sublethally irradiated (6.5Gy) congenic recipients. After 5–6 weeks, AML-bearing mice were treated with vehicle or 22d by intraperitoneal injection (50mg/kg/dose) twice a day for 2 weeks (Figure 6A). We found that Ac-p53 and Ac-SMC3 levels were effectively induced as early as 2 days after initiating 22d treatment (Figure 6B). Post-treatment analysis revealed a significantly reduced frequency (p=0.0097) and number (p=0.0101) of dTomato+/cKit+ AML cells in the BM and spleen of 22d-treated compared to vehicle-treated mice (Figure 6C–E, not shown). To further assess LSC activity, we transplanted BM cells (2 × 106) from these treated mice into secondary recipients. A significantly reduced frequency (p<0.0001) and number (p=0.0006) of dTomato+/cKit+ AML cells were observed in the BM at 8 weeks (Figure 6F, G), along with a significantly reduced spleen weight and AML infiltration (Figure 6H, not shown). We monitored leukemia onset and survival in a cohort of secondary recipients who received BM cells (5×106) from 22d- or vehicle-treated mice. While all vehicle-treated transplants (n=5) succumbed to aggressive AML with a median survival of 51 days, only 2 (of 4) 22d-treated transplants showed signs of leukemia with prolonged latency (median survival 270 days; Figure 6I). Collectively, these results indicate that in vivo 22d treatment effectively diminishes AML burden and eliminates the leukemia-initiating capacity of LSCs in CM AML mouse model.
Figure 6. Administration of HDAC8i 22d in vivo effectively reduces AML burden and abrogates LSC activity.

A. Schematic illustration of the experimental design. CM/tdTomato+ AML cells (2×106) were transplanted into cohorts of congenic mice. After 5–6 weeks, mice were treated with vehicle or 22d (50mg/kg/dose) twice a day for 2 weeks. AML engraftment was analyzed at the end of treatment and BM cells were transplanted into second recipients, which were analyzed for engraftment at 8 weeks or monitored for leukemia onset and survival monitored up to 1 year.
B. Western blot analysis of Ac-p53, Ac-SMC3 levels in BM cells from mice treated with HDAC8i 22d or vehicle.
C. Representative FACS plots showing gating and frequency of dTomato+/cKit+ AML cells in the BM of vehicle- (top) or 22d (bottom)-treated mice.
D. Frequency of AML cells in the BM of mice treated with vehicle (n=13) or 22d (n=13) for 2 weeks. Each dot represents results from an individual mouse and lines indicate mean ± SEM. **P=0.0097
E. Total number of AML cells in the BM of mice treated with vehicle (n=13) or 22d (n=13) for 2 weeks. *P=0.01
F. Frequency of AML cells in the BM of secondary transplant recipients of BM from vehicle- (n=5) or 22d-treated (n=4) mice. ***P<0.0001
G. Total number of AML cells in the BM of second transplant recipients of vehicle (n=5) or 22d treated (n=4) BM. ***P=0.0006
H. Spleen weight of second transplant recipients of vehicle (n=5) or 22d treated (n=4) BM. *P=0.0159
I. Survival curve of second transplant recipients of vehicle- (n=5) or 22d-treated (n=4) BM. **P=0.0049
To further evaluate the efficacy of targeting human AML LSCs, we expanded primary patient-derived inv(16)+ AML cells in NOD/SCID/IL-2R-γ−/−/Tg (CMV-IL3, CSF2, KITLG) mice (NSGS). T-cell depleted AML cells (1×106) were transplanted into irradiated (300cGy) NSGS mice via intrafemoral injection. Human AML (hCD45+) cells were selected from leukemic BM for transplantation into larger cohorts. When levels of hCD45+ AML cells reached 5–6% in sampled BM aspirates (Figure S6A), we began treatment with 22d or vehicle twice daily for 2 weeks (Figure 7A). Two of the six vehicle-treated mice were moribund with aggressive leukemia before the end of treatment while all 22d-treated mice (n=5) survived. AML burden indicated by the frequency and numbers of hCD45+ AML cells and hCD45+/CD34+ AML stem/progenitor cells were significantly lowered in BM, PB and spleen of 22d-treated compared to vehicle-treated mice (Figure 7B–F, S6B–C). No significant change in immunophenotype was seen for the remaining hCD45+ cells (Figure S6D, E). We further assessed the effects on LSC leukemia-initiating activity by transplanting BM cells (4 × 106) from 22d- or vehicle-treated mice. While significant AML burden (29–41% hCD45+; 19.5–26.8% hCD45+/CD34+) was observed in vehicle control transplants at 12 weeks, we found only minimal residual hCD45+ cells (0.63–1.08%) and hCD45+/CD34+ AML stem/progenitor cells (0.28–0.46%) in 22d-treated secondary transplants (Figure 7G–I, S6F–I). The frequencies of primitive CD34+ population within remaining hCD45+ cells were significantly reduced in recipients of 22d-treated BM (Figure 7J), suggesting a selective loss of AML stem/progenitor cells. In line with a depletion of AML LSCs by HDAC8i, we did not find any reoccurrence of AML in 22d treated secondary transplants compared to lethal regrowth of AML (median survival 93 days) in all vehicle treated transplants (Figure 7K). Altogether, these results provide compelling evidence that in vivo administration of HDAC8i 22d profoundly abrogates AML propagation and eliminates long-term maintenance of functional LSCs.
Figure 7. Propagation of primary inv(16)+ AML is diminished by in vivo HDAC8i 22d treatment.

A. Schematic illustration of the experimental design. Primary inv(16)+ AML cells from patient AML1070 (see Table S1) were depleted of T cells and directly injected intrafemorally into irradiated (300cGy) NSGS mice (1×106 cells/mouse). Human AML hCD45+ cells were selected from the leukemic BM for transplantation (2×106 hCD45+ cells/mouse) into larger cohorts of NSGS mice. When AML progression is evident, we began treatment with 22d (50mg/kg/dose) or vehicle twice daily for 2 weeks. Treated mice were then analyzed for AML burden and BM cells were transplanted into secondary recipients.
B. Representative FACS plots showing gating and frequency of hCD45+ (left) and hCD45+/CD34+ (right) cells in mice received vehicle (top) or 22d (bottom) treatment for 2 weeks.
C. Frequency of hCD45+ AML cells in the PB of vehicle or 22d treated mice (n=5; P=0.0159).
D. Frequency of hCD45+ AML cells in the BM of vehicle or 22d treated mice (n=5; P=0.0079).
E. Frequency of hCD45+/CD34+ AML cells in the BM of vehicle or 22d treated mice (n=5; P=0.0079).
F. The spleen weight of mice treated with vehicle (n=5) or 22d (n=5). *P=0.0159
G. Representative FACS plots of hCD45+/CD34+ AML cell frequency in BM of vehicle- or 22d-treated secondary transplants 12 weeks after transplantation.
H. Frequency of hCD45+ AML cells in BM of vehicle- or 22d-treated secondary transplants (n=4; P=0.0007).
I. Frequency of hCD45+/CD34+ AML cells in BM of vehicle- or 22d-treated secondary transplants (n=4; P=0.0006).
J. Frequency of immunophenotypic populations within hCD45+ cells in BM of vehicle- or 22d-treated secondary transplants (n=4; *P<0.05; ***P<0.001).
K. Survival curve of 2nd transplant recipients of vehicle- (n=4) or 22d-treated (n=3) BM up to 5 months after transplantation. *P=0.0285
See also Figure S6.
Discussion
In this study, we delineate a leukemogenic mechanism whereby the CM fusion protein disrupts p53 activity through aberrant post-translational modification mediated by HDAC8. We demonstrate that genetic deletion of Hdac8 or inhibition of HDAC8 activity drastically prevented LSC transformation and maintenance. These studies indicate that HDAC8 is a valuable target for further therapeutic development in inv(16)+ AML and potentially other HDAC8-overexpressing cancers.
The molecular basis of CM’s leukemogenic activity has been debated over the years. Dominant inhibition of RUNX proteins by CM, either through cytoplasmic sequestration (Adya et al., 1998; Kanno et al., 1998) or constitutive repression (Lutterbach et al., 1999; Durst et al., 2003), was conventionally viewed as the main leukemogenic mechanism. However, several recent studies revealed that CM-induced leukemogenesis in fact requires functional RUNX proteins rather than RUNX inhibition. First, we showed that high Runx2 expression facilitates CM-induced AML transformation (Kuo et al., 2009). Second, the high affinity RUNX1 binding site presumably underlying dominant RUNX inhibition (Lukasik et al., 2002) seemed dispensable for CM-leukemogenesis (Kamikubo et al., 2010). Third, RUNX1 activity was reported required for CM leukemogenesis and growth of CBF AML cells (Hyde et al., 2015; Ben-Ami et al., 2013; Goyama et al., 2013). Here, we propose a model that HDAC8-mediated deacetylation of p53 is a major mechanism contributing to CM-leukemogenesis. Although RUNX1 is not required for CM-p53 interaction, RUNX-binding could possibly facilitate localization of CM in the nucleus where CM-p53 interaction is detected. The C-terminal deletion CM-ΔC95 does not bind HDAC8 or impair p53 response, consistent with its lack of leukemogenic activity (Kamikubo et al., 2012). We found that CM residues 179-221, located at the SMMHC fusion junction, are required for binding to p53. This region may contribute to direct p53 binding or the unique conformational properties of the CM fusion protein. Overall, our study highlights that the CM fusion protein gains additional leukemogenic functions beyond its role in RUNX inhibition.
We found that HDAC8 mRNA expression is selectively higher (5–12 fold) in inv(16)+ CD34+ AML cells compared to non-inv(16) AML or normal CD34+ cells. Overall, non-inv(16) AML CD34+ cells also express higher levels (2–3 fold on average) of HDAC8 compared to normal CD34+ cells. This could possibly explain the higher sensitivity to HDAC8i 22d observed for non-inv(16) AML CD34+ relative to normal CD34+. The mechanism underlying this activity needs to be further defined, but these results raise the possibility that HDAC8i might be applicable to additional subsets of AML patients. It is not surprising that t(8;21)+ AML is also somewhat sensitive to HDAC8i given that RUNX1-ETO fusion protein is known to interact with repressor complexes and HDACs (Amann et al., 2001). In fact, anti-leukemia activities in t(8;21)+ AML have been described for class I HDAC inhibitors (Yang et al., 2007; Odenike et al., 2008; Bots et al., 2014). In inv(16)+ AML, the HDAC8 level is approximately 2-fold higher in CD34+ population relative to CD34− cells in each patient (Figure S4E). However, no significant correlation was seen between HDAC8 levels and sensitivity to HDAC8i 22d (Figure S4F). We also did not observe any difference in sensitivity for samples with additional common mutations such as c-KIT mutation (Figure S4G). Our results suggest that given the CM-p53-HDAC8 protein complex, inv(16)+ cells are particularly sensitive to HDAC8i regardless of HDAC8 levels. Notably, HDAC8i treatment enhances the chemosensitivity of inv(16)+ CD34+ cells, highlighting its potential efficacy in overcoming chemotherapy resistance. Importantly, the apoptotic effect of HDAC8i is selective for LSCs compared to normal HSCs, which display little increase in p53 activity and normal engraftment capacity after HDAC8i treatment. This selectivity is likely due to the combination of oncogenic stress and the recruitment of HDAC8 and p53 into a stable protein complex in inv(16)+ cells. HDAC8 was recently shown to repress Runx2 activity and osteogenic differentiation (Fu et al., 2014). Therefore, additional microenvironmental factors could possibly contribute to the in vivo functional effects of HDAC8i.
Given that TP53 is rarely mutated in inv(16)+ AML, our results not only elucidate an alternative oncogenic lesion-specific p53-inactivating mechanism, but also highlight a unique therapeutic opportunity to selectively modulate p53 activity in inv(16)+ AML. Inhibition of MDM2 by Nutlins or other inhibitors has been explored as an approach to activate p53 in AML with varying efficacy (Kojima et al., 2005; Long et al., 2010) underscoring the need to further dissect the heterogeneity and oncogene-specific mechanisms. HDAC1, 2, 3 and Sirtuins are known to modulate p53 activity by deacetylation (Juan et al., 2000; Luo et al., 2000; Ito et al., 2002). We show that HDAC8 can deacetylate p53 and modulate its activity, similar to other class I HDACs. Inhibiting HDAC8 substantially enhances p53 activation and diminishes AML cell survival under oncogenic or genotoxic stress, supporting HDAC8 as a promising therapeutic target. Most HDAC inhibitors including vorinostat, currently used or being tested in clinical trials display relatively low activity against HDAC8 (Khan et al., 2008). Our team has recently developed ortho-aryl N-hydroxycinnamides including 22d (Huang et al., 2012) with superior anti-HDAC8 selectivity and potency compared to the hydroxamic acid inhibitor PCI-34051 previously reported (Balasubramanian et al., 2008). All HDAC8i compounds we tested showed similar effects as HDAC8 knock-down or deletion. Compared to other class I-, III- or broad-spectrum HDAC inhibitors, 22d selectively enhanced Ac-p53 and the HDAC8-specific substrate Ac-SMC3. Further, the sensitivity to 22d can be modulated by genetically manipulating HDAC8 or p53 status. These results indicate that a 22d regimen, at least at lower doses, reactivates p53 through specific inhibition of HDAC8. Inhibition of other HDAC members or other off-target effects is possible at higher doses (e.g., >20 uM). Clearly, further development and improvement of HDAC8i for pre-clinical and clinical studies is warranted.
In conclusion, we demonstrate a mechanism whereby CM aberrantly interacts with p53 and HDAC8, leading to deacetylation and inactivation of p53 by HDAC8. Our results indicate that HDAC8 acts to promote CM-associated AML LSC transformation and maintenance. Remarkably, HDAC8i treatment restores p53 acetylation and activity, induces apoptosis, and abrogates AML propagation and the leukemia-initiating activity of inv(16)+ LSCs. This study highlights HDAC8 as an attractive therapeutic target and provides the rationale for designing HDAC8-directed therapies to enhance eradication of inv(16)+ AML LSCs in combination with chemotherapy.
Experimental Procedures
Human Samples
Mobilized PBSC were obtained from healthy donors and AML samples were obtained from patients (Table S1) at City of Hope (COH). CD34+ cell isolation was performed using MACS or CliniMACS (Miltenyi Biotech, Germany). Samples were acquired with signed informed consent and approval by the COH Institutional Review Board, in accordance with an assurance filed with and approved by the Department of Health and Human Services.
Mice
All Cbfb56M/+ (Kuo et al., 2006) and Mx1-Cre (Kühn et al., 1995) mice used were backcrossed to C57BL/6 for more than 10 generations. C57BL/6, Ai14 Cre reporter and NSGS mice were obtained from the Jackson Laboratory. To induce CM, 6–8 week old Mx1-Cre/Cbfb56M/+ mice were intraperitoneally (i.p.) injected with 250 μg of pIpC (InvivoGen) every other day for 14 days (7 doses). Similarly treated Cbfb56M/+ littermates were used as control. Pre-leukemic cells were isolated 2 weeks after the last dose of pIpC. AML transplant was performed via tail vein injection into sub-lethally irradiated (6.5 Gy) 6–8-week-old congenic C57BL/6 mice (CD45.1+). Human AML cells were depleted of T cells using MACS (Miltenyi Biotech) and transplanted into irradiated (300cGy) NSGS mice via intrafemoral injection. In vivo 22d/vehicle treatment was performed by i.p. injection (50mg/kg/dose) twice a day. All mice were maintained in an AAALAC-accredited animal facility, and all procedures were performed in accordance with federal and state government guidelines and established institutional guidelines and protocols approved by the Institutional Animal Care and Use Committee at the Beckman Research Institute of COH.
Immunoprecipitation (IP) and Western Blotting
Cells were lysed in RIPA buffer containing a protease inhibitor cocktail (Roche) and MG132. Antibodies used for IP were conjugated with protein A/G beads using the antibody cross-linking kit (Pierce Biotechnology, Rockford, IL) following the manufacturer’s instructions. For western blot analysis, proteins were resolved using 10% SDS-PAGE. The antibodies used included anti-FLAG (M2; Sigma), anti-HDAC8 (H145, E5; Santa Cruz), anti-CBFβ (141,4,1; Santa Cruz), anti-p53 (DO-1, 1C12), anti-Ac-p53 (K379) (Cell Signaling, Danvers, MA), anti-SMC3 (Bethyl Laboratories), anti-Ac-SMC3 (MBL International) and anti-β-actin (Sigma, St. Louis, MO). Horseradish peroxidase–conjugated anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch, West Grove PA) were used, followed by detection using the SuperFemto kit (Pierce Biotechnology, Rockford, IL).
Statistics
Statistical analyses were performed with Student’s t test or analysis of variance (ANOVA) for normal distributions. Mann-Whitney U tests were used when the criteria for a normal distribution were not satisfied. A p value less than 0.05 was considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Supplementary Material
Highlights.
CBFβ-SMMHC (CM) forms an aberrant protein complex with p53 and HDAC8
HDAC8 promotes CM-mediated LSC transformation by aberrantly deacetylating p53
HDAC8 inhibition selectively targets inv(16)+ AML CD34+ cells by reactivating p53
Inhibition of HDAC8 eliminates AML propagation and LSC leukemia-initiating activity
Acknowledgments
This work was supported by the V Foundation Scholar Award (to Y.-H.K.), the American Cancer Society (123278-RSG-12-140-01-CSM to Y.-H.K.), the National Cancer Institute grants R01 CA178387 (to Y.-H.K.) and P30 CA033572 (to COH). J.Q. is supported by California Institute of Regenerative Medicine training grant TG2-01150 (to COH). We thank Dr. Pual Liu (NIH) for the generous gift of CM deletion plasmids; Dr. Gerard Evan (U of Cambridge) for the p53ERTAM KI mice; Dr. Mu-Shui Dai (Oregon Health & Sciences University) for the p53 reporter plasmid; Dr. Wei Gu (Columbia University) for the p53-WT and p53-8KR plasmids; and Dr. Sriram Balasubramanian (Pharmacyclics) for providing PCI-34051. We thank the excellent technical support of the Analytical Cytometry Core and Animal Resource Center at COH.
Footnotes
Author Contributions:
J.Q., S.S. performed research, analyzed data, participated in manuscript writing; W-K. H., Q.C., S-W.C., L.L. performed research, reviewed the manuscript; H.L., Y.H., T.M., A.L. performed research, reviewed the manuscript; G.M., R.B., W.-J. H., C.-I.C designed research, interpreted data, reviewed the manuscript; Y-H.K. designed research, analyzed and interpreted data, and wrote the manuscript.
The authors declared that no conflicts of interest exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adya N, Stacy T, Speck NA, Liu PP. The leukemic protein core binding factor beta (CBFβ)-smooth-muscle myosin heavy chain sequesters CBFalpha2 into cytoskeletal filaments and aggregates. Mol Cell Biol. 1998;18:7432–443. doi: 10.1128/mcb.18.12.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, Downing JR, Meyers S, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–483. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, Lotem J, Tanay A, Groner Y. Addiction of t(8;21) and inv(16) Acute Myeloid Leukemia to Native RUNX1. Cell Rep. 2013;4:1131–143. doi: 10.1016/j.celrep.2013.08.020. [DOI] [PubMed] [Google Scholar]
- Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, Stanley K, Shortt J, Ossenkoppele GJ, et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. 2014;123:1341–352. doi: 10.1182/blood-2013-03-488114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla LH, Wijmenga C, Wang Q, Stacy T, Speck NA, Eckhaus M, Marín-Padilla M, Collins FS, Wynshaw-Boris A, Liu PP. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87:687–696. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Martin-Zanca D, Soucek L, Lawlor ER, Brown-Swigart L, Verschuren EW, Evan GI. Temporal dissection of p53 function in vitro and in vivo. Nat Genet. 2005;37:718–726. doi: 10.1038/ng1572. [DOI] [PubMed] [Google Scholar]
- Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal Protein L23 Activates p53 by Inhibiting MDM2 Function in Response to Ribosomal Perturbation but Not to Translation Inhibition. Mol Cell Biol. 2004;24:7654–668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–17. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol. 2003;23:607–619. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhang P, Ge J, Cheng J, Dong W, Yuan H, Du Y, Yang M, Sun R, Jiang H. Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone H3K9 acetylation and RUNX2 activity. Int J Biochem Cell Biol. 2014;54:68–77. doi: 10.1016/j.biocel.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, Nakagawa M, Olsson A, Wunderlich M, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123:3876–888. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22:1539–541. doi: 10.1038/leu.2008.143. [DOI] [PubMed] [Google Scholar]
- Huang WJ, Wang YC, Chao SW, Yang CY, Chen LC, Lin MH, Hou WC, Chen MY, Lee TL, et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. Chem Med Chem. 2012;7:1815–824. doi: 10.1002/cmdc.201200300. [DOI] [PubMed] [Google Scholar]
- Hyde RK, Zhao L, Alemu L, Liu PP. Runx1 is required for hematopoietic defects and leukemogenesis in Cbfb-MYH11 knock-in mice. Leukemia. 2015 doi: 10.1038/leu.2015.58. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, Wu CW. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. 2000;275:20436–443. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- Kamikubo Y, Hyde RK, Zhao L, Alemu L, Rivas C, Garrett LJ, Liu PP. The C-terminus of CBFβ-SMMHC is required to induce embryonic hematopoietic defects and leukemogenesis. Blood. 2012;121:638–642. doi: 10.1182/blood-2012-06-434688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikubo Y, Zhao L, Wunderlich M, Corpora T, Hyde RK, Paul TA, Kundu M, Garrett L, Compton S, et al. Accelerated leukemogenesis by truncated CBFβ-SMMHC defective in high-affinity binding with RUNX1. Cancer Cell. 2010;17:455–468. doi: 10.1016/j.ccr.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Kanno T, Sakakura C, Bae SC, Ito Y. Cytoplasmic sequestration of the polyomavirus enhancer binding protein 2 (PEBP2)/core binding factor alpha (CBFalpha) subunit by the leukemia-related PEBP2/CBFβ-SMMHC fusion protein inhibits PEBP2/CBF-mediated transactivation. Mol Cell Biol. 1998;18:4252–261. doi: 10.1128/mcb.18.7.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–89. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106:3150–59. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YH, Gerstein RM, Castilla LH. Cbfβ-SMMHC impairs differentiation of common lymphoid progenitors and reveals an essential role for RUNX in early B-cell development. Blood. 2008;111:1543–551. doi: 10.1182/blood-2007-07-104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, Le Beau MM, Kogan SC, Castilla LH. Cbfβ-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006;9:57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Kuo YH, Zaidi SK, Gornostaeva S, Komori T, Stein GS, Castilla LH. Runx2 induces acute myeloid leukemia in cooperation with Cbfβ-SMMHC in mice. Blood. 2009;113:3323–332. doi: 10.1182/blood-2008-06-162248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–29. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Liu P, Tarlé SA, Hajra A, Claxton DF, Marlton P, Freedman M, Siciliano MJ, Collins FS. Fusion between transcription factor CBFβ/PEBP2β and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–44. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- Liu PP, Wijmenga C, Hajra A, Blake TB, Kelley CA, Adelstein RS, Bagg A, Rector J, Cotelingam J, et al. Identification of the chimeric protein product of the CBFB-MYH11 fusion gene in inv(16) leukemia cells. Genes Chromosomes Cancer. 1996;16:77–87. doi: 10.1002/(SICI)1098-2264(199606)16:2<77::AID-GCC1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Long J, Parkin B, Ouillette P, Bixby D, Shedden K, Erba H, Wang S, Malek SN. Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood. 2010;116:71–80. doi: 10.1182/blood-2010-01-261628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- Lukasik SM, Zhang L, Corpora T, Tomanicek S, Li Y, Kundu M, Hartman K, Liu PP, Laue TM, et al. Altered affinity of CBFβ-SMMHC for Runx1 explains its role in leukemogenesis. Nat Struct Biol. 2002;9:674–79. doi: 10.1038/nsb831. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Hou Y, Durst KL, Hiebert SW. The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A. 1999;96:12822–27. doi: 10.1073/pnas.96.22.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno DA, Scrideli CA, Cortez MAA, de Paula Queiroz R, Valera ET, da Silva Silveira V, Yunes JA, Brandalise SR, Tone LG. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150:665–673. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- Odenike OM, Alkan S, Sher D, Godwin JE, Huo D, Brandt SJ, Green M, Xie J, Zhang Y, et al. Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clin Cancer Res. 2008;14:7095–7101. doi: 10.1158/1078-0432.CCR-08-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–30. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Prébet T, Boissel N, Reutenauer S, Thomas X, Delaunay J, Cahn JY, Pigneux A, Quesnel B, Witz F, et al. Acute myeloid leukemia with translocation (8;21) or inversion (16) in elderly patients treated with conventional chemotherapy: a collaborative study of the French CBF-AML intergroup. J Clin Oncol. 2009;27:4747–753. doi: 10.1200/JCO.2008.21.0674. [DOI] [PubMed] [Google Scholar]
- Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- Ustun C, Marcucci G. Emerging diagnostic and therapeutic approaches in core binding factor acute myeloid leukaemia. Curr Opin Hematol. 2015;22:85–91. doi: 10.1097/MOH.0000000000000124. [DOI] [PubMed] [Google Scholar]
- Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, Bushweller JH, Bories JC, Alt FW, et al. The CBFβ subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- Yang G, Thompson MA, Brandt SJ, Hiebert SW. Histone deacetylase inhibitors induce the degradation of the t(8;21) fusion oncoprotein. Oncogene. 2007;26:91–101. doi: 10.1038/sj.onc.1209760. [DOI] [PubMed] [Google Scholar]
- Zhao L, Cannons JL, Anderson S, Kirby M, Xu L, Castilla LH, Schwartzberg PL, Bosselut R, Liu PP. CBFB-MYH11 hinders early T-cell development and induces massive cell death in the thymus. Blood. 2007;109:3432–440. doi: 10.1182/blood-2006-10-051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389–1402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.