

Abstract
A key event in the generation of a cellular response against malicious organisms through the endocytic pathway is binding of peptidic antigens by major histocompatibility complex class II (MHC class II) molecules. The bound peptide is then presented on the cell surface where it can be recognized by T helper lymphocytes. NetMHCIIpan is a state-of-the-art method for the quantitative prediction of peptide binding to any human or mouse MHC class II molecule of known sequence. In this paper, we describe an updated version of the method with improved peptide binding register identification. Binding register prediction is concerned with determining the minimal core region of nine residues directly in contact with the MHC binding cleft, a crucial piece of information both for the identification and design of CD4+ T cell antigens. When applied to a set of 51 crystal structures of peptide-MHC complexes with known binding registers, the new method NetMHCIIpan-3.1 significantly outperformed the earlier 3.0 version. We illustrate the impact of accurate binding core identification for the interpretation of T cell cross-reactivity using tetramer double staining with a CMV epitope and its variants mapped to the epitope binding core. NetMHCIIpan is publicly available at http://www.cbs.dtu.dk/services/NetMHCIIpan-3.1.
Keywords: MHC class II, Peptide binding, Tcell cross-reactivity, Binding core, Artificial neural networks, Peptide-MHC
Introduction
Major histocompatibility complex class II (MHC class II) molecules play an essential role in the cellular immune system of vertebrates. The main function of MHC class II molecules consists of loading short peptide fragments derived from exogenously derived antigenic proteins and presenting them on the antigen presenting cell surface, where they can be recognized by T helper lymphocytes. If the peptide fragment is of foreign origin, the T cells can help initiating an appropriate immune response (Castellino et al. 1997; Germain 1994; Rudolph et al. 2006).
A key characteristic of T cells is that they are antigen specific, but also cross-reactive (Wilson et al. 2004). Specificity is an indicator of the ability of T cells to discriminate between different antigens, a crucial property that allows the immune system to distinguish between self and non-self material. As the number of potential peptidic antigens is much larger than the T cell receptor (TCR) repertoire diversity, it appears inevitable that a single T cell should have the ability to recognize multiple peptide-MHC complexes (Birnbaum et al. 2014; Sewell 2012). This degeneracy in T cell recognition is commonly referred to as cross-reactivity, and has been implicated both in immune protection and disease (Benoist and Mathis 2001; Lang et al. 2002; Welsh et al. 2010). The selection of T cell epitopes is primarily driven by the delicate balance between specificity and binding degeneracy of both the MHC and the TCR. While the contribution of the MHC in the selection of antigens has to a high degree been described and explained, the role of the T cell receptor remains an essential missing link in our understanding of Tcell immune responses.
Because the peptide-binding groove of MHC class II molecules is open at both ends, there are limited constraints on the length of the peptide ligand, which can protrude out at both ends of the pocket. Although normally only about 9 amino acids of the peptide, the so-called binding core, are directly interacting with residues of the MHC groove, peptides of up to 30 amino acids (even whole proteins) can be loaded onto MHC class II molecules (Chicz et al. 1993; Sette et al. 1989). Peptide-MHC binding affinity is largely determined by the primary amino acid sequence of the peptide-binding core. However, it has been shown that the peptide flanking regions (PFRs) on either side of the binding core can affect peptide-MHC binding and, ultimately, immunogenicity (Carson et al. 1997; Godkin et al. 2001). Human MHC class II molecules (called HLA class II, here abbreviated HLA-II) are highly polymorphic, comprising thousands of different allelic variants across the population. HLA-II binding motifs are generally rather degenerate, and promiscuous peptides with the ability to bind to several alleles have been identified (Al-Attiyah and Mustafa 2004; Sturniolo et al. 1999). Promiscuous peptides can either share the same anchors across different alleles, or contain overlapping binding cores with allele-specific anchors.
Given the critical role of MHC class II in the selection of peptides for antigen presentation and immune response orchestration, large efforts have been dedicated to the development of high-throughput methods for the screening of peptide binding to MHC class II. Although significant progress has been made toward developing cost-effective experimental methods for screening of peptide binding to MHC class II (exemplified by Justesen et al. (2009)), the cost of performing an exhaustive characterization of the binding specificity of all prevalent MHC class II molecules remains prohibitive.
Computational methods for the prediction of MHC class II binding, an attractive alternative to costly experimental methods, have evolved steadily in the past years. They include ARB (Bui et al. 2005), SVRMHC (Wan et al. 2006), MHCpred (Doytchinova and Flower 2003), NetMHCII (Nielsen and Lund 2009), TEPITOPE (Sturniolo et al. 1999), and a limited number of pan-specific methods covering also molecules for which scarce or no measured binding data are available, including TEPITOPEpan (Zhang et al. 2012) and NetMHCIIpan-3.0 (Karosiene et al. 2013). With variable degrees of accuracy, all these methods allow the identification of peptides that are likely binders of MHC class II molecules. However, when it comes to identification of the MHC binding core, most of these methods have limited predictive performance (Zhang et al. 2012). The current version of NetMHCIIpan (version 3.0) achieves a higher performance than TEPITOPEpan in terms of predicted binding affinity; however, it is less accurate for the task of identifying the correct binding core (Zhang et al. 2012).
The NetMHCIIpan method is based on an ensemble of artificial neural networks trained on quantitative peptide binding data covering multiple MHC class II molecules. Ensembles are in general superior to individual networks because the selection of the networks weights is an optimization problem with many local minima (Hansen and Salamon 1990). However, although most networks in the ensemble may pick up the salient characteristics distinguishing binders from non-binders in terms of amino acid preferences and binding anchors, they often disagree on the precise location of the minimal 9-mer core residues interacting with the MHC cleft. We have previously shown (Andreatta et al. 2011) that the identification of the binding core by neural network ensembles can be greatly improved with the employment of a network alignment procedure called “offset correction”. This method is fully automated, and unsupervised. This means that no information about the actual location of the binding core is used to define the offset values.
In this paper, we apply offset correction to the NetMHCIIpan network ensemble to enhance MHC class II binding core recognition. Besides accurately identifying the binding core, the method assigns reliability scores to each binding core prediction and allows the quantification of the likelihood of multiple binding cores within a single antigenic peptide. Using tetramer double staining with a CMV epitope and its variants, we illustrate the importance of reliable binding core identification for the interpretation of T cell recognition and cross-reactivity.
Materials and methods
Data sets
The method was trained on data used in the original NetMHCIIpan-3.0 publication (data available at http://www.cbs.dtu.dk/suppl/immunology/NetMHCIIpan-3.0). This set consists of quantitative peptide-MHC class II binding data from the Immune Epitope Database (Vita et al. 2015). It comprises 52,062 affinity measurements covering 24 HLA-DR, 5 HLA-DP, 6 HLA-DQ, and 2 murine H-2 molecules. The IC50 (half inhibitory concentration) values in nM were log-transformed using the formula 1-log(IC50)/log(50,000) as described by Nielsen et al. (2003) to fall in the range between 0 and 1. Additionally, a set of 9860 binding affinity measurements covering 13 HLA-DR alleles introduced by Karosiene et al. (2013) was used as an independent evaluation set.
For the binding core benchmark, we compiled a list of 51 crystal structures of peptide-MHC class II complexes from the PDB database (Rose et al. 2015). They comprise 36 HLA-DR, 6 HLA-DQ, 5 HLA-DP, and 4 H-2 structures with a bound peptide in their binding cleft. The minimal 9-mer cores were manually annotated by pinpointing in the 3-D structures the peptide residues in contact with the MHC anchor pockets (typically positions P1, P4, P6, and P9, depending on the allele).
Neural network architecture and training
The input sequences were presented to the input layer of each network as described by Nielsen et al. (2008), using only BLOSUM encoding, where each amino acid is encoded as the BLOSUM50 matrix score vector of 20 amino acids (Henikoff and Henikoff 1992). The optimal 9-mer core of a peptide therefore required 9×20=180 input neurons. Forty additional input neurons were used to encode the composition of the peptide flanking regions (PFRs), calculated as the average BLOSUM scores on a maximum window of three amino acids at either end of the binding core (Nielsen et al. 2008). C- and N-terminal PFR lengths (LPFR) were each encoded using two input neurons with values LPFR/(LPFR+1) and 1-LPFR/(LPFR+1) respectively. The peptide length L was encoded with two input neurons taking the values LPEP and 1-LPEP, where LPEP=1/(1+exp((L-15)/2)). These transformations ensure that the normalized input values to the neural networks fall in the range between 0 and 1. MHC molecules were represented in terms of a pseudo-sequence defined by polymorphic residues in potential contact with a bound peptide (Nielsen et al. 2007a). We used the same pseudosequences of 34 residues for the MHC alpha and beta chains defined by Karosiene et al. (2013), resulting in additional 34× 20=680 inputs. As a result, the total size of the input layer amounted to 906 neurons.
The ensemble of artificial neural networks was trained as described by Karosiene et al. (2013), using a fivefold cross-validation; alternative hidden layers of 10, 15, 40, and 60 hidden neurons; and 10 initial configurations of the network weights for each architecture. Starting from an initial random weight configuration, the networks were trained in an iterative manner by predicting the strongest binding 9-mer core for each training sequence and subsequently minimizing the difference between its predicted and measured binding affinity. No experimental information about the location of the binding core was used in the model construction. This procedure was repeated multiple times using different initial random configurations to construct the multiple networks that constitute the NetMHCIIpan ensemble. The resulting complete ensemble was composed of 200 networks. Predictions of binding affinity were then calculated as the ensemble average, and binding cores as the majority vote (for details see Nielsen et al. (2010)). In order to minimize over-estimation of predictive performance, the subsets for cross-validation were generated using the procedure described by Nielsen et al. (2007b), which clusters together peptides that share identical stretches of at least nine amino acids.
NetMHCIIpan-3.1 was trained on the same data set and with a nearly identical architecture to the previous version NetMHCIIpan-3.0. The only difference affecting the prediction binding affinity is the encoding of the PFR lengths to correct for a small inconsistency in the previous version of the method, which truncated the right but not the left PFR to a maximum of three amino acids. Note, that offset correction does not affect the prediction of binding affinity. In a fivefold cross-validation experiment on the 37 molecules included in the training set, the performances of NetMHCIIpan-3.1 and 3.0 were not significantly different, with an average area under the ROC curve (AUC) of 0.870 for the former and 0.871 for the latter. The AUC value weighed by the number of sequences per allele was 0.871 for both versions. Similarly, on an independent test set comprising additional 9860 peptide-MHC complexes not included in the training, the predictive performances of versions 3.0 and 3.1 are not significantly different and both reach an averaged AUC=0.808 and weighed AUC=0.807.
Calculation of offsets
Offset correction, introduced by Andreatta et al. (2011), is a procedure that allows combining the binding core prediction of multiple neural networks in an ensemble. The sequence motif identified by each network is first represented as a position-specific scoring matrix (PSSM), storing the background-corrected frequency of each amino acid at each position in the binding core. Next, using Gibbs sampling, the PSSMs are aligned to generate an average PSSM with highest Kullback-Leibler distance (KLD) from the background amino acid frequency in natural proteins. The extent of the shift to the left or to the right produced by the alignment for each PSSM (and its relative network) is the “offset” value associated to that given network. When the prediction for a peptide is made, the optimal binding core of each network is shifted according to its relative offset value. This procedure, previously shown to improve the identification of binding motifs of individual HLA-DP and DQ molecules (Andreatta and Nielsen 2012), is here generalized for the pan-specific MHC class II binding problem.
Core reliability scores
We define the core reliability score as the fraction of networks in the ensemble that agrees on a given binding core register. The complete profile of reliability scores for a peptide-MHC pair is referred to as the core histogram, and the optimal core selected by NetMHCIIpan-3.1 is the core with highest reliability score (i.e., the majority vote).
Peptide binding to HLA class II
Peptide-HLA class II binding affinities were determined as previously described (Justesen et al. 2009). Briefly, denatured and purified recombinant HLA class II α- and β-chains were diluted into a refolding buffer containing graded concentrations of the test peptide, and incubated for 48 h at 18 °C to allow for equilibrium to be reached.
Complex formation was detected using a proximity-based luminescent oxygen channeling immunoassay and the peptide concentration leading to half-saturation (ED50) was determined as previously described (Justesen et al. 2009). Under the limited receptor concentrations used here, the ED50 reflects the affinity of the interaction.
HLA class II tetramers
HLA class II tetramers were produced as previously described. Briefly, recombinant α- and β-chains (HLA-DRA1 and DRB5*01:01) were folded in the presence of either the wild-type IE1211-225 peptide, P3 variant, or P7 variant of the wild-type peptide. The resulting monomers were tetramerized with PE- or APC-conjugated streptavidin.
PBMC's from a donor previously determined to recognize the DRB5*01:01 restricted CMV IE1211–225 epitope (Braendstrup et al. 2014) was expanded on this epitope. The cells were double-stained with PE-labeled wild-type IE1211–225 -DRB5*01:01 tetramer and APC-labeled mutant peptide-DRB5*01:01 tetramer as previously described (Braendstrup et al. 2013). The cells were washed and subsequently stained with anti-CD3-Pacific blue and anti-CD4-PerCP antibody (Biolegend, San Diego, USA) for 30 min. The stained cells were analyzed by flow cytometry on a Fortessa (BD Biosciences).
Results
Accurate prediction of the peptide-binding core register
The main innovation in NetMHCIIpan-3.1 is the introduction of offset correction to improve the identification of the peptide binding core register. We compiled a list of 51 crystal structures of peptide-MHC class II complexes from PDB (Rose et al. 2015), inspecting the location of the bound peptide core within the MHC binding groove. Note that only one of these peptide-MHC pairs is present in the training set and only ten additional peptides are present with extended residues either at the N or C terminal. However, since the location of the binding core is never used as training information (learning is uniquely based on the affinity values), evaluating the predictive performance in terms of correctly identified binding cores remains entirely independent.
Several predictors were applied to this set of peptides-MHC structures to identify the location of the peptide binding cores. On the 36 peptides in complex with HLA-DR alleles, we made binding core predictions using NetMHCII-2.2 (Nielsen et al. 2007b), NetMHCIIpan-3.0 (Karosiene et al. 2013), and TEPITOPEpan (Zhang et al. 2012). NetMHCIIpan-3.1 identified correctly 33 out of 36 binding core registers, compared to 26 correct cores for NetMHCIIpan-3.0 and 32 correct cores for TEPITOPEpan (see Table 1). NetMHCII-2.2 is not a pan-specific method and only covers a subset of the alleles in the benchmark. On the subset of 31 peptide-MHCs covered by NetMHCII-2.2, the core was predicted correctly in 20 cases. On this subset of peptides, NetMHCIIpan-3.1 recognizes the correct binding core in 28/31 instances. The comparison to NetMHCIIpan-3.0 and NetMHCII-2.2 is in both cases statistically significant (p<0.01, comparison of ratios).
Table 1. Prediction of peptide binding core registers for 36 peptide/HLA-DR complexes from PDB.
| PDB | Allele | Antigen | Core (PDB) | NetMHCII | NetMHCIIpan-3.0 | TEPITOPEpan | NetMHCIIpan-3.1 | Reliability |
|---|---|---|---|---|---|---|---|---|
| 2FSE | DRB1*01:01 | AGFKGEQGPKGEPG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG | 0.910 |
| 1J8H | DRB1*04:01 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | 0.910 |
| 1FYT | DRB1*01:01 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | 0.905 |
| 3L6F | DRB1*01:01 | APPAYEKLSAEQSPP | YEKLSAEQS | YEKLSAEQS | YEKLSAEQS | YEKLSAEQS | YEKLSAEQS | 0.900 |
| 2Q6W | DRB3*01:01 | AWRSDEALPLGS | WRSDEALPL | WRSDEALPL | WRSDEALPL | WRSDEALPL | WRSDEALPL | 0.900 |
| 1A6A | DRB1*03:01 | PVSKMRMATPLLMQA | MRMATPLLM | MRMATPLLM | MRMATPLLM | MRMATPLLM | MRMATPLLM | 0.890 |
| 2IPK | DRB1*01:01 | XPKWVKQNTLKLAT | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL | 0.875 |
| 1SJH | DRB1*01:01 | PEVIPMFSALSEG | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | 0.870 |
| 4H1L | DRB3*03:01 | QHIRCNIPKRISA | IRCNIPKRI | IRCNIPKRI | IRCNIPKRI | IRCNIPKRI | 0.855 | |
| 3QXA | DRB1*01:01 | PVSKMRMATPLLMQA | MRMATPLLM | KMRMATPLL | MRMATPLLM | MRMATPLLM | MRMATPLLM | 0.840 |
| 3PGD | DRB1*01:01 | KMRMATPLLMQALPM | MRMATPLLM | KMRMATPLL | MRMATPLLM | MRMATPLLM | MRMATPLLM | 0.835 |
| 3PDO | DRB1*01:01 | KPVSKMRMATPLLMQALPM | MRMATPLLM | KMRMATPLL | MRMATPLLM | MRMATPLLM | MRMATPLLM | 0.835 |
| 1AQD | DRB1*01:01 | VGSDWRFLRGYHQYA | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ | 0.830 |
| 1PYW | DRB1*01:01 | XFVKQNAAALX | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL | 0.820 |
| 1KLG | DRB1*01:01 | GELIGTLNAAKVPAD | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | 0.805 |
| 3C5J | DRB3*03:01 | QVIILNHPGQISA | IILNHPGQI | IILNHPGQI | VIILNHPGQ | IILNHPGQI | 0.780 | |
| 4H26 | DRB3*03:01 | QWIRVNIPKRI | IRVNIPKRI | IRVNIPKRI | IRVNIPKRI | IRVNIPKRI | 0.760 | |
| 4OV5 | DRB1*01:01 | GSDARFLRGYHLYA | ARFLRGYHL | ARFLRGYHL | ARFLRGYHL | FLRGYHLYA | ARFLRGYHL | 0.705 |
| 4IS6 | DRB1*04:01 | WNRQLYPEWTEAQRLD | LYPEWTEAQ | LYPEWTEAQ | LYPEWTEAQ | LYPEWTEAQ | LYPEWTEAQ | 0.690 |
| 4H25 | DRB3*03:01 | QHIRCNIPKRIGPSKVATLVPR | IRCNIPKRI | IGPSKVATL | IRCNIPKRI | IRCNIPKRI | 0.675 | |
| 1SJE | DRB1*01:01 | PEVIPMFSALSEGATP | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | 0.655 |
| 1H15 | DRB5*01:01 | GGVYHFVKKHVHES | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH | 0.620 |
| 1T5X | DRB1*01:01 | AAYSDQATPLLLSPR | YSDQATPLL | YSDQATPLL | YSDQATPLL | YSDQATPLL | YSDQATPLL | 0.615 |
| 1BX2 | DRB1*15:01 | ENPVVHFFKNIVTPR | VHFFKNIVT | VVHFFKNIV | VVHFFKNIV | VHFFKNIVT | VHFFKNIVT | 0.615 |
| 4MD4 | DRB1*04:01 | ATEYRVRVNSAYQDK | YRVRVNSAY | YRVRVNSAY | YRVRVNSAY | YRVRVNSAY | YRVRVNSAY | 0.605 |
| 2SEB | DRB1*04:01 | AYMRADAAAGGA | MRADAAAGG | YMRADAAAG | YMRADAAAG | YMRADAAAG | YMRADAAAG | 0.595 |
| 1ZGL | DRB5*01:01 | VHFFKNIVTPRTPGG | FKNIVTPRT | FFKNIVTPR | FFKNIVTPR | FKNIVTPRT | FKNIVTPRT | 0.570 |
| 4I5B | DRB1*01:01 | VVKQNCLKLATK | VVKQNCLKL | VKQNCLKLA | VVKQNCLKL | VVKQNCLKL | VKQNCLKLA | 0.560 |
| 4MD5 | DRB1*04:04 | SAVRLRSSVPGVR | VRLRSSVPG | VRLRSSVPG | AVRLRSSVP | VRLRSSVPG | VRLRSSVPG | 0.555 |
| 1HQR | DRB5*01:01 | VHFFKNIVTPRTP | FKNIVTPRT | FFKNIVTPR | FFKNIVTPR | FKNIVTPRT | FKNIVTPRT | 0.555 |
| 4MCZ | DRB1*04:01 | GVYATRSSAVRLR | YATRSSAVR | YATRSSAVR | VYATRSSAV | VYATRSSAV | VYATRSSAV | 0.515 |
| 4MCY | DRB1*04:01 | SAVRLRSSVPGVR | VRLRSSVPG | VRLRSSVPG | VRLRSSVPG | VRLRSSVPG | VRLRSSVPG | 0.510 |
| 1FV1 | DRB5*01:01 | NPVVHFFKNIVTPRTPPPSQ | FKNIVTPRT | FFKNIVTPR | FFKNIVTPR | FKNIVTPRT | FKNIVTPRT | 0.480 |
| 1YMM | DRB1*15:01 | ENPVVHFFKNIVTPRGGSGGGGG | VHFFKNIVT | VVHFFKNIV | VVHFFKNIV | VHFFKNIVT | VHFFKNIVT | 0.460 |
| 4MDI | DRB1*04:02 | SAVRLRSSVPGVR | VRLRSSVPG | VRLRSSVPG | VRLRSSVPG | VRLRSSVPG | 0.395 | |
| 4AEN | DRB1*01:01 | MPLAQMLLPTAMRMKM | MLLPTAMRM | LAQMLLPTA | QMLLPTAMR | MLLPTAMRM | MLLPTAMRM | 0.315 |
|
| ||||||||
| Correct: | 20/31 | 26/36 | 32/36 | 33/36 | ||||
Core (PDB) is the validated binding register as observed in the PDB crystal structures. Results are sorted by decreasing reliability score, and incorrect predictions are highlighted in grey
On a set of 15 peptides in complex with HLA-DP, HLA-DQ, and H-2 molecules, NetMHCIIpan-3.1 predicted the correct binding core in 12 cases, whereas NetMHCIIpan-3.0 was correct in 9 cases (Table 2). TEPITOPEpan is limited to HLA-DR and cannot produce predictions for these molecules.
Table 2. Prediction of peptide binding core registers for 14 peptides in complex with HLA-DP, HLA-DQ, and H-2 molecules from PDB.
| PDB | Allele | Antigen | Core (PDB) | NetMHCIIpan-3.0 | NetMHCIIpan-3.1 | Reliability |
|---|---|---|---|---|---|---|
| 1JK8 | DQA1*03:03-DQB1*03:02 | LVEALYLVCGERGG | EALYLVCGE | EALYLVCGE | EALYLVCGE | 0.685 |
| 1S9V | DQA1*05:05-DQB1*02:01 | LQPFPQPELPY | PFPQPELPY | LQPFPQPEL | LQPFPQPEL | 0.505 |
| 1UVQ | DQA1*01:02-DQB1*06:02 | MNLPSTKVSWAAVGGGGSLV | LPSTKVSWA | TKVSWAAVG | TKVSWAAVG | 0.440 |
| 4GG6 | DQA1*03:01-DQB1*03:02 | QQYPSGEGSFQPSQENPQ | EGSFQPSQE | EGSFQPSQE | EGSFQPSQE | 0.420 |
| 4D8P | DQA1*03:01-DQB1*02:01 | PQPEQPEQPFPQP | EQPEQPFPQ | EQPEQPFPQ | EQPEQPFPQ | 0.340 |
| 4OZG | DQA1*05:05-DQB1*02:01 | APQPELPYPQPGS | PQPELPYPQ | PQPELPYPQ | PQPELPYPQ | 0.300 |
|
| ||||||
| 4P4K | DPA1*01:03-DPB1*02:01 | QAFWIDLFETIG | FWIDLFETI | FWIDLFETI | FWIDLFETI | 0.650 |
| 4P57 | DPA1*01:03-DPB1*02:01 | QAFWIDLFETIGGGSLV | FWIDLFETI | FWIDLFETI | FWIDLFETI | 0.630 |
| 3LQZ | DPA1*01:03-DPB1*02:01 | RKFHYLPFLPSTGGS | FHYLPFLPS | RKFHYLPFL | FHYLPFLPS | 0.625 |
| 3WEX | DPA1*02:01-DPB1*05:01 | KVTVAFNQFGGS | KVTVAFNQF | KVTVAFNQF | VAFNQFGGS | 0.450 |
| 4P5M | DPA1*01:03-DPB1*02:01 | QAYDGKDYIALKG | YDGKDYIAL | YDGKDYIAL | YDGKDYIAL | 0.425 |
|
| ||||||
| 1MUJ | H-2-IAb | PVSKMRMATPLLMQA | MRMATPLLM | MRMATPLLM | MRMATPLLM | 0.725 |
| 4P23 | H-2-IAb | FEAQKAKANKAVD | AQKAKANKA | FEAQKAKAN | AQKAKANKA | 0.475 |
| 1IAO | H-2-IAd | ISQAVHAAHAEI | SQAVHAAHA | ISQAVHAAH | SQAVHAAHA | 0.475 |
| 2IAD | H-2-IAd | HATQGVTAASSHE | TQGVTAASS | HATQGVTAA | TQGVTAASS | 0.420 |
|
| ||||||
| Correct: | 9/15 | 12/15 | ||||
Core (PDB) is the validated binding register as observed in the PDB crystal structures. Results are sorted by decreasing reliability score, and incorrect predictions are highlighted in grey
Offset values are conserved within different MHC loci
Offset values for the 200 networks in the ensemble were calculated for all HLA-DR, HLA-DP, HLA-DQ, and H-2 molecules in the training set. Figure 1 shows the cumulative distribution of the number of networks that have assigned the same offset value on at least x% molecules in the same locus or on all molecules, where x is the value on the x-axis. For instance, the same offset value was found across the 24 HLA-DR molecules (100 % agreement) for 135 networks, whereas 180 networks have at least 75 % offset agreement, and all 200 networks have the same offset for at least 50 % of the HLA-DR molecules. Observing that offset values to a high degree are conserved across alleles in a given locus (more than 50 % of the networks agree on all alleles in a given locus) but not across loci (rhombus series in Fig. 1), we used a majority vote scheme to compile a separate list of offset values for each locus HLA-DR, DP, DQ, and H-2. The advantage of a single list of offsets per locus, as opposed to offset values for each molecule, is that it can be applied in a pan-specific manner to other alleles in the same locus that were not included in the training set. Besides covering a comprehensive list of over 5000 MHC class II molecules to choose from, the method can handle custom MHC amino acid sequences. In this case, the list of offset values calculated over all isotypes is used.
Fig. 1.
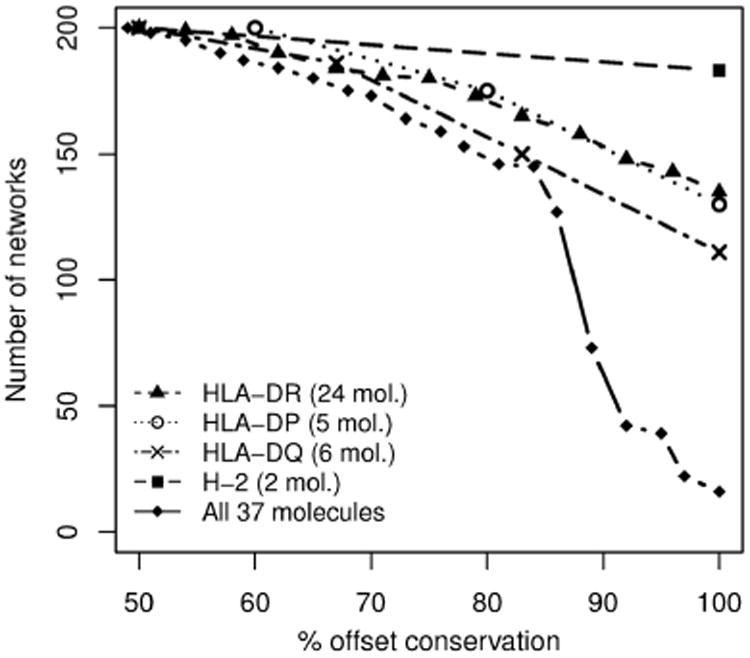
Offset value conservation across MHC alleles. The plot shows a cumulative distribution of the number of networks (out of the 200 in the ensemble) that were assigned the same offset value on at least x% molecules. Within the same locus, the offset values are highly conserved, whereas there is more discordance across loci (rhombus series with continuous line)
Reliability scores profiles
The output of the NetMHCIIpan-3.1 server comprises graphical profiles of the core reliability scores of binding peptides. Score profiles offer a probabilistic representation of the location of the binding core by displaying the fraction of networks in the neural network ensemble that select any given binding register. In cases where the networks identify a clear and unequivocal binding core, the reliability score profile will show a single peak at the P1 of the predicted binding core. For example, the peptide PVSKMRMATPLLMQA comprised in the PDB benchmark (Table 1) is predicted to be a strong binder to HLA-DRB1*03:01 with a predicted affinity of 11 nM. Most networks (89 %) place the P1 of the binding core at position 4 in the peptide, predicting the 9-mer core to be MRMATPLLM (Fig. 2a). In other cases, the binding core is more degenerate. For instance, the peptide NPVVHFFKNIVTPRTPPPSQ is predicted to be a strong binder to the molecule HLA-DRB5*01:01 (predicted affinity = 22 nM), but its reliability core profile shows two possible binding cores each with reliability score >0.4 (Fig. 2b). The core FKNIVTPRT identified by the NetMHCIIpan-3.1 is the correct register as seen in the PDB crystal structure 1FV1; however, also the shifted version FFKNIVTPR satisfies well the anchor requirements for DRB5*01:01 and is suggested by the profile as an alternative binding register. Toggling the reliability profile graphical option in the NetMHCIIpan-3.1 server submission page generates such profiles for any submitted sequence.
Fig. 2.
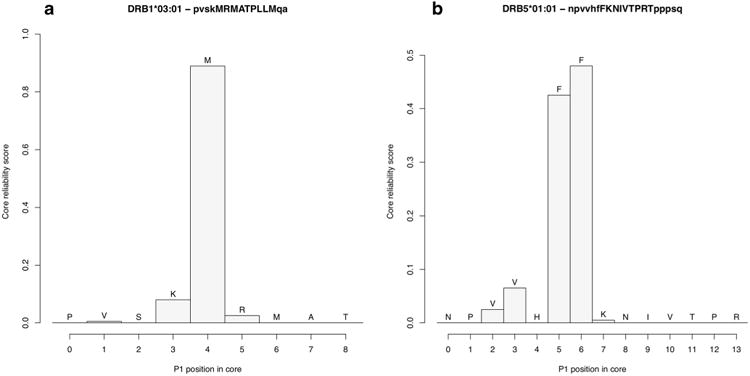
Core reliability profiles for two peptide-MHC complexes in the PDB benchmark. The x-axis shows the location of the first position (P1) of the 9-mer core within the peptide, also highlighted with uppercase characters on top of the plot. The height of each bar is the core reliability score assigned by NetMHCIIpan-3.1 to each alternative core register
Reliability scores correlate with predicted binding core correctness
As discussed in the methods, we defined a reliability score assigned to each core prediction as the fraction of networks in an ensemble that select a given binding core register. NetMHCIIpan-3.1 produces a reliability score for the optimal register of each predicted binder as part of its prediction. For the 51 peptide-MHCs in Tables 1 and 2, sorted by reliability scores, we observe that false core prediction tend to fall in the lower part of the list (i.e., they have lower reliability scores). In particular, with a threshold on the reliability score = 0.6 (at least 60 % of the networks in the ensemble agreeing on the best binding core), we obtain 30 peptides with correctly predicted cores without a single false positive.
Generally, HLA-DQ molecules appeared to have lower reliability scores than molecules from different isotypes (both in the cases exemplified in Table 2 and in further predictions not shown here). A previous characterization of the binding motifs of HLA-DQ molecules (Andreatta and Nielsen 2012) showed rather degenerate motifs and less-defined binding anchors compared to HLA-DR and HLA-DP. Our results confirm this observation and suggest a more promiscuous binding mode over alternative binding cores for HLA-DQ.
Interpretation of Tcell cross-reactivity guided by accurate core identification
To illustrate how accurate identification of the binding core of MHC class II ligands can aid the interpretation of Tcell cross-reactivity, we generated a set of five variants of the CMV epitope IE1211–225 (NIEFFTKNSAFPKTT) restricted to HLA-DRB5*01:01 (Braendstrup et al. 2014). The peptide-binding core of the WT peptide was identified using the NetMHCIIpan-3.1 method. Then, we introduced targeted mutations at the primary P1 anchor position and at two additional non-anchor MHC positions. The complete set of peptide variants is listed in Table 3.
Table. 3. Targeted mutations to a CMV epitope and their impact on MHC binding and T cell cross-reactivity.
| Peptide | Mutation | IC50 | Rank (%) | Core reliability | Measured IC50 (nM) | % Cross-reactivity |
|---|---|---|---|---|---|---|
| NIEFFTKNSAFPKTT | WT | 7.19 | 0.40 | 0.818 | 14 | 100 |
| NIEFETKNSAFPKTT | P1 F→E | 656 | 50.0 | 0.400 | 173 | NB |
| NIEFFTRNSAFPKTT | P3 K→R | 6.52 | 0.30 | 0.855 | 7 | 94 |
| NIEFFTYNSAFPKTT | P3 K→Y | 7.37 | 0.40 | 0.815 | 8 | 3 |
| NIEFFTKNSAYPKTT | P7 F→Y | 8.71 | 0.80 | 0.800 | 12 | 97 |
| NIEFFTKNSARPKTT | P7 F→R | 6.77 | 0.40 | 0.800 | 20 | 41 |
The data set contains a wild-type CD4 epitope for the molecule HLA-DRB5*01:01 obtained from Braendstrup et al. (2014), and five variants constructed as described in the text. The predicted binding core for the WT peptide is underlined, mutations are highlighted in bold. Conservative mutations were defined as having a positive Blosum62 score, and non-conservative mutations as having a negative Blosum62 score. Predicted IC50, Rank and Core reliability values were obtained using the NetMHCIIpan-3.1 method. Measured IC50 was obtained as described in methods. % Cross-reactivity is the percent of CD4 Tcells specific for the WT tetramer that also share specificity of the peptide variant. NB indicates that no tetramer formation was detected
From these peptide variations and corresponding binding affinity and binding core predictions, we would expect the P1 variant to lose binding to the HLA molecule, whereas the effect on MHC binding for all the P3 and P7 variants should be minimal. In terms of T cell cross-reactivity, we would predict that the P3 and P7 variants with conservative mutations would be cross-reactive with T cells raised against the WT peptide, whereas the P3 and P7 variants with non-conservative mutations would not (Frankild et al. 2008). In order to validate these predictions, we made tetramers of the six peptides (WT and variants) and tested for cross-reactivity of the variants to the WT peptide (Fig. 3).
Fig. 3.
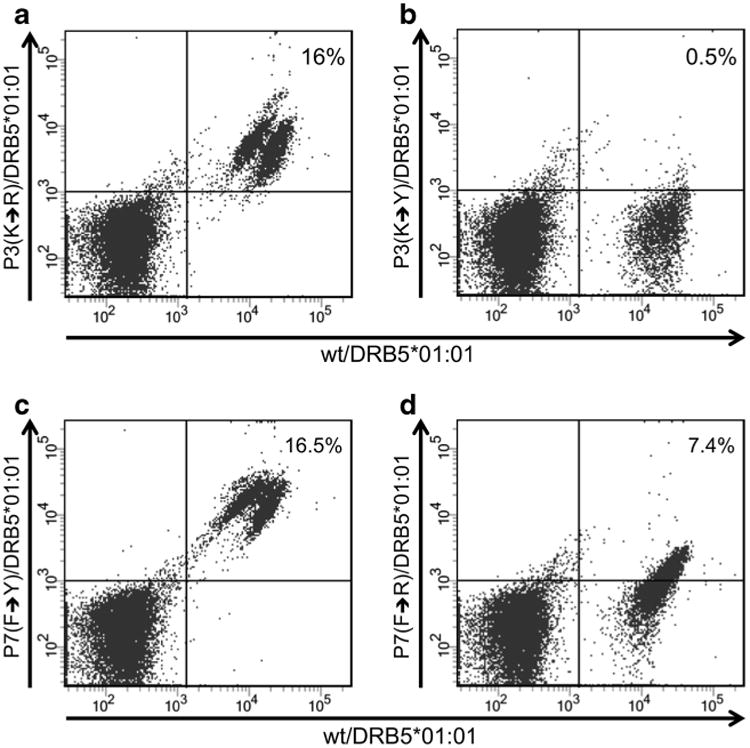
T cell cross-recognition between P3 and P7 variants and the wild-type epitope IE1211–225. T cells were expanded for 12 days on the wild-type peptide IE1211– 225 peptide (wt). The specific T cells were double stained with in all cases PE-labeled IE1211–225 / DRB5*01:01 together within a APC-labeled P3(K→R)variant/ DRB5*01:01 tetramer; in b APC-labeled P3(K→Y)variant/ DRB5*01:01 tetramer; in c APC-labeled P7(F→Y)variant/ DRB5*01:01 tetramer; in d APC-labeled P7(F→R)variant/ DRB5*01:01. The cells were subsequently stained for anti-CD3 and -CD4. The plots show gated CD4+ T cells, and the frequency of tetramer+ CD4+T cells is indicated
In all five cases, the tetramer and binding affinity measurements confirmed our predictions. The P1 variant had lost its binding to the HLA-DRB5*01:01 molecule, and no tetramers could be formed. The two conservative P3 and P7 variants showed close to 100 % cross-reactivity to the WT epitope, and the non-conservative variants displayed a significant loss in cross-reactivity to the WT in both cases.
Discussion
Prediction of peptide-MHC class II binding involves answering two questions: does the peptide bind to the MHC, and if so, where does it bind. Addressing the first question, prediction of binding affinity for MHC class II has reached, depending on the molecule of interest and method employed, performance values between 0.80 and 0.90 in terms of AUC. In particular, the state-of-the-art method NetMHCIIpan-3.0 (Karosiene et al. 2013) can produce accurate binding predictions for any HLA-DR, DP, DQ, and H-2 molecule of known sequence, and the prediction values can readily be interpreted in terms of IC50 binding affinity.
However, when it comes to the second question, we have shown here that NetMHCIIpan-3.0 often fails to identify the correct binding register. As the peptide-binding groove of MHC class II molecules is open at both ends, long peptide ligands can potentially bind with alternative 9-residue core sequences forming interactions with the MHC binding pockets. It is important to stress that the neural networks of NetMHCIIpan are only trained on affinity data and not on the binding registers, which are rather learned as a by-product of the procedure. Because the individual networks constituting the NetMHCIIpan ensemble of 200 networks are trained on different subsets of the data set and alternative configurations, they may disagree on the placement of the binding core register, leaving us with the problem of combining their predictions.
In the method presented in this paper, we applied “offset correction,” a procedure that allows combining the predictions of different neural networks in an ensemble, to obtain improved core register identification with unaltered binding affinity performance. When employed on a set of 51 crystal structures of peptide-MHC complexes with known binding register, the new method NetMHCIIpan-3.1 identified the correct core register in 45 cases, improving from the 35 correct predictions of NetMHCIIpan-3.0. Besides achieving a higher performance in terms of predicted binding affinity than TEPITOPEpan (Zhang et al. 2012), NetMHCIIpan-3.1 also has comparable accuracy in terms of predicted binding cores, and can be applied on a wider range of MHC class II molecules. Using this improved method, we illustrate the importance of accurate binding core identification for the interpretation of T cell cross-reactivity using tetramer double staining with a CMV epitope and selected variants defined with respect to the epitope binding core.
Precise identification of the peptide binding register is imperative for the fine characterization of the MHC class II binding pocket, both for the discovery and design of pep-tides with the ability to give rise to MHC recognition. Altering peptide-MHC anchors does in most cases abolish binding, whereas mutations in other positions that are not directly in contact with the binding core can often be accepted without losing binding to the MHC (Anderson and Gorski 2003). In cases where cross-reactivity occurs, pep-tides with mutated non-anchor amino acids can still be recognized by the TCR and stimulate an immune response (Basu et al. 2000). In addition to this, several studies demonstrated the relationship between the MHC binding core and patterns of TCR recognition (Arnold et al. 2002; Bremel and Homan 2014). Thus, reliable binding core identification could facilitate identification of TCR recognition motifs for CD4+ T cell epitopes.
In summary, we showed that NetMHCIIpan-3.1 is the state-of-the-art both for the quantitative prediction of binding affinity and the identification of the peptide binding core register. The prediction program is publicly available as a convenient and easy-to-use web server at http://www.cbs.dtu.dk/services/NetMHCIIpan-3.1.
Acknowledgments
Funding This work was supported with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201200010C, and from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2012-0115). MN is a researcher at the Argentinean national research council (CONICET).
References
- Al-Attiyah R, Mustafa AS. Computer-assisted prediction of HLA-DR binding and experimental analysis for human promiscuous Th1- cell peptides in the 24 kDa secreted lipoprotein (LppX) of Mycobacterium tuberculosis. Scand J Immunol. 2004;59:16–24. doi: 10.1111/j.0300-9475.2004.01349.x. [DOI] [PubMed] [Google Scholar]
- Anderson MW, Gorski J. Cutting edge: TCR contacts as anchors: effects on affinity and HLA-DM stability. J Immunol. 2003;171:5683–5687. doi: 10.4049/jimmunol.171.11.5683. [DOI] [PubMed] [Google Scholar]
- Andreatta M, Nielsen M. Characterizing the binding motifs of 11 common human HLA-DP and HLA-DQ molecules using NNAlign. Immunology. 2012;136:306–311. doi: 10.1111/j.1365-2567.2012.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M, Schafer-Nielsen C, Lund O, et al. NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS One. 2011;6:e26781. doi: 10.1371/journal.pone.0026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold PY, La Gruta NL, Miller T, et al. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J Immunol. 2002;169:739–749. doi: 10.4049/jimmunol.169.2.739. [DOI] [PubMed] [Google Scholar]
- Basu D, Horvath S, Matsumoto I, et al. Molecular basis for recognition of an arthritic peptide and a foreign epitope on distinct MHC molecules by a single TCR. J Immunol. 2000;164:5788–5796. doi: 10.4049/jimmunol.164.11.5788. [DOI] [PubMed] [Google Scholar]
- Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- Birnbaum ME, Mendoza JL, Sethi DK, et al. Deconstructing the peptide-MHC specificity of Tcell recognition. Cell. 2014;157:1073–1087. doi: 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braendstrup P, Justesen S, Osterbye T, et al. MHC class II tetramers made from isolated recombinant α and β chains refolded with affinity-tagged peptides. PLoS One. 2013;8:e73648. doi: 10.1371/journal.pone.0073648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braendstrup P, Mortensen BK, Justesen S, et al. Identification and HLA-tetramer-validation of human CD4+ and CD8+ T cell responses against HCMV proteins IE1 and IE2. PLoS One. 2014;9:e94892. doi: 10.1371/journal.pone.0094892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremel RD, Homan EJ. Frequency patterns of T-cell exposed amino acid motifs in immunoglobulin heavy chain peptides presented by MHCs. Front Immunol. 2014;5:541. doi: 10.3389/fimmu.2014.00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Peters B, et al. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–314. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]
- Carson RT, Vignali KM, Woodland DL, Vignali DA. Tcell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity. 1997;7:387–399. doi: 10.1016/s1074-7613(00)80360-x. [DOI] [PubMed] [Google Scholar]
- Castellino F, Zhong G, Germain RN. Antigen presentation by MHC class II molecules: invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum Immunol. 1997;54:159–169. doi: 10.1016/s0198-8859(97)00078-5. [DOI] [PubMed] [Google Scholar]
- Chicz RM, Urban RG, Gorga JC, et al. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. Towards the in silico identification of class II restricted T-cell epitopes: a partial least squares iterative self-consistent algorithm for affinity prediction. Bioinformatics. 2003;19:2263–2270. doi: 10.1093/bioinformatics/btg312. [DOI] [PubMed] [Google Scholar]
- Frankild S, de Boer RJ, Lund O, et al. Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS One. 2008;3:e1831. doi: 10.1371/journal.pone.0001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- Godkin AJ, Smith KJ, Willis A, et al. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J Immunol. 2001;166:6720–6727. doi: 10.4049/jimmunol.166.11.6720. [DOI] [PubMed] [Google Scholar]
- Hansen LK, Salamon P. Neural network ensembles. IEEE Trans Pattern Anal Mach Intell. 1990;12:993–1001. doi: 10.1109/34.58871. [DOI] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justesen S, Harndahl M, Lamberth K, et al. Functional recombinant MHC class II molecules and high-throughput peptide-binding as-says. Immunome Res. 2009;5:2. doi: 10.1186/1745-7580-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karosiene E, Rasmussen M, Blicher T, et al. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–724. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang HLE, Jacobsen H, Ikemizu S, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinf. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007a;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinf. 2007b;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, et al. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol. 2008;4:e1000107. doi: 10.1371/journal.pcbi.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Justesen S, Lund O, et al. NetMHCIIpan-2.0—improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 2010;6:9. doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PW, Prlić A, Bi C, et al. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- Sette A, Adorini L, Colon SM, et al. Capacity of intact proteins to bind to MHC class II molecules. J Immunol. 1989;143:1265–1267. [PubMed] [Google Scholar]
- Sewell AK. Why must Tcells be cross-reactive? Nat Rev Immunol. 2012;12:669–677. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturniolo T, Bono E, Ding J, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Liu W, Xu Q, et al. SVRMHC prediction server for MHC-binding peptides. BMC Bioinf. 2006;7:463. doi: 10.1186/1471-2105-7-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev. 2010;235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DB, Wilson DH, Schroder K, et al. Specificity and degeneracy of T cells. Mol Immunol. 2004;40:1047–1055. doi: 10.1016/j.molimm.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen Y, Wong HS, et al. TEPITOPEpan: extending TEPITOPE for peptide binding prediction covering over 700 HLA-DR molecules. PLoS One. 2012;7:e30483. doi: 10.1371/journal.pone.0030483. [DOI] [PMC free article] [PubMed] [Google Scholar]