

Abstract
Regulation of L-type calcium current is critical for the development, function, and regulation of many cell types. CaV1.2 channels that conduct L-type calcium currents are regulated by many protein kinases, but the sites of action of these kinases remain unknown in most cases. We combined mass spectrometry (LC-MS/MS) and whole-cell patch clamp techniques in order to identify sites of phosphorylation of CaVβ subunits in vivo and test the impact of mutations of those sites on CaV1.2 channel function in vitro. Using the CaV1.1 channel purified from rabbit skeletal muscle as a substrate for phosphoproteomic analysis, we found that Ser193 and Thr205 in the HOOK domain of CaVβ1a subunits were both phosphorylated in vivo. Ser193 is located in a potential consensus sequence for casein kinase II, but it was not phosphorylated in vitro by that kinase. In contrast, Thr205 is located in a consensus sequence for cAMP-dependent phosphorylation, and it was robustly phosphorylated in vitro by PKA. These two sites are conserved in multiple CaVβ subunit isoforms, including the principal CaVβ subunit of cardiac CaV1.2 channels, CaVβ2b. In order to assess potential modulatory effects of phosphorylation at these sites separately from effects of phosphorylation of the α11.2 subunit, we inserted phosphomimetic or phosphoinhibitory mutations in CaVβ2b and analyzed their effects on CaV1.2 channel function in transfected nonmuscle cells. The phosphomimetic mutation CaVβ2bS152E decreased peak channel currents and shifted the voltage dependence of both activation and inactivation to more positive membrane potentials. The phosphoinhibitory mutation CaVβ2bS152A had opposite effects. There were no differences in peak CaV1.2 currents or voltage dependence between the phosphomimetic mutation CaVβ2bT164D and the phosphoinhibitory mutation CaVβ2bT164A. However, calcium-dependent inactivation was significantly increased for the phosphomimetic mutation CaVβ2bT164D. This effect was subunit-specific, as the corresponding mutation in the palmitoylated isoform, CaVβ2a, had no effect. Overall, our data identify two sites of conserved phosphorylation of the HOOK domain of CaVβ subunits in vivo and reveal differential modulatory effects of phosphomimetic mutations in these sites. These results reveal a new dimension of regulation of CaV1.2 channels through phosphorylation of the HOOK domains of their β subunits.
Keywords: L-type Ca2+ channel, Ventricular Myocytes, Electrophysiology
1. Introduction
Protein kinases regulate L-type calcium currents conducted by voltage-gated (CaV) channels to precisely control numerous physiological functions of nerve and muscle cells. CaV1.1 and CaV1.2 channels are regulated by a number of protein kinases (PKA, CaMKII, AKT/PKB, PKC and PKG) [1, 2]. PKA phosphorylation is required for up-regulating L-type calcium currents through CaV1.1 and CaV1.2 channels in the fight-or-flight response [2-5], and CaMKII is essential for frequency-dependent potentiation [6]. Dysregulation of protein kinases is implicated in many pathological conditions [7-9]. Despite its importance in physiology and pathophysiology, the molecular mechanisms of regulation of CaV1.1 and CaV1.2 channels by protein phosphorylation remain incompletely understood.
CaV1.1 and CaV1.2 channels in nerve and muscle cells consist of a pore-forming α1 subunit in association with CaVβ, CaVα2δ, and possibly CaVγ subunits [2, 10, 11]. Calmodulin (CaM), a ubiquitous Ca binding protein, is also associated with this protein complex [12]. CaVα1 subunits are composed of four homologous domains (I-IV) with six transmembrane segments (S1-S6) and a reentrant pore loop in each [2]. Multiple regulatory sites are located in the large C-terminal domain of CaV1.1 and CaV1.2 channels [13-16], which is subject to in vivo proteolytic processing near its center [13, 17-19]. An IQ motif in the proximal C-terminus is implicated in Ca/calmodulin-dependent inactivation [14, 15]. Noncovalent interaction of the distal C-terminus with the proximal C-terminal domain has an auto-inhibitory effect by reducing coupling efficiency of gating charge movement to channel opening [16, 20, 21], and the proximal C-terminus EF-hand is required to mediate the auto-inhibitory effect of the distal C-terminus [22]. Recently, it was shown that this autoinhibitory CaV1.2 signaling complex with an A Kinase Anchoring Protein bound is sufficient to recapitulate the stimulatory actions of PKA on CaV1.2 channels in a non-muscle cell system [23]. This reconstituted regulatory system has allowed functional tests of the role of phosphorylation sites in the α1 subunits in calcium channel regulation.
In our previous studies, we took advantage of the ease of purification of CaV1.1 channels from rabbit skeletal muscle to identify sites of in vivo phosphorylation of the α1 subunits [24]. We then used our reconstituted regulatory system to analyze the functional effects of mutations in the homologous sites in the CaV1.2 channel, which are highly conserved. We found that two conserved sites located at the interface between the distal and proximal C-terminal domains were required for regulation of basal and PKA-stimulated channel activity [23]. Both a PKA site at Ser1700 and a casein kinase II site at Thr1704 were required for normal regulation of basal channel activity, whereas only Ser1700 was required for stimulation of channel activity by PKA [23]. These results suggest that PKA phosphorylation of CaV1.2 at Ser1700 relieves the autoinhibition of the distal C-terminal on CaV1.2 channel function allowing the PKA-dependent increase in current amplitude. Mice with mutations in Ser1700 and Thr1704 have greatly reduced basal L-type calcium currents and much reduced response to β-adrenergic stimulation [25, 26], as expected from these studies in transfected nonmuscle cells.
Phosphorylation sites in CaVβ subunits were identified previously by a variety of biochemical and proteomic techniques [18, 27-29], but the level of phosphorylation in vivo, and the physiological significance of these phosphorylation sites remain uncertain. In the experiments described here, we have used mass spectrometry (LC-MS/MS) of purified skeletal muscle CaV1.1 channels for phosphoproteomic analysis and whole-cell patch clamp studies of the expressed CaV1.2 channel for functional analysis. With this approach, we identified two in-vivo phosphorylation sites in the Hook domain of CaVβ subunits. Phosphomimetic and phosphoinhibitory mutations in the analogous sites differentially modulate the function of full-length CaV1.2 channels expressed in nonmuscle cells.
2. Experimental Procedures
2.1. Protein purification, and sample preparation
All animal procedures were conducted in compliance with the recommendations of the Institutional Animal Care and Use Committee of the University of Washington. Rabbit skeletal muscle CaV1.1 channels were purified as previously described [30]. Male New Zealand White rabbits (2.5 kg, Western Oregon Rabbit Co) were euthanized by lethal injection of pentobarbital, skeletal muscle was rapidly harvested, rinsed briefly in phosphate buffered saline (PBS, pH 7.4), snap frozen in liquid nitrogen, and stored at −80°C. All purification steps were carried out at 4°C in the presence of the following protease and phosphatase inhibitors: 1 μM aprotinin, 1 μM pepstatin, 10 μM leupeptin, 100 μM benzamidine, 1 mM phenanthroline, 10 μM E-64, 20 μg/μL soybean trypsin inhibitor, 0.5 mM NaVO4, 100 nM microcystin, 10 nM cyclosporin A. CaV1.1 was purified using wheat germ agglutinin linked agarose (Vector Labs) and anion exchange DEAE Sephadex A-25 (GE Healthcare), chromatography [30, 31]. Pure protein was snap frozen in liquid nitrogen and stored at −80°C in single-use aliquots. For mass spectrometric analysis, 1-2.5 μg of pure protein was separated by SDS-PAGE, the band of interest was excised and subjected to in-gel trypsin (Gold, Promega) digestion by standard procedures (http://donatello.ucsf.edu/ingel.html); 1-3 pmol of tryptic peptides were analyzed by mass spectrometry.
2.2. In vitro phosphorylation
Pure CaV1.1 was first dephosphorylated with 5 U Calf Intestinal-alkaline Phosphatase (CIP, NEB) for 2 h at 30°C in assay buffer (20 mM Tris pH7.5, 10 mM MgCl2, 2 mM DTT, 10 mM NaCl, 200 mM ATP) and phosphatase was quenched with 10 mM NaVO4. Then CaV1.1 was phosphorylated with 100 U PKA (Sigma, from Bovine heart), 100 U CaMKII (NEB), or 25 U CK2 (NEB) for 2.5 h at 30°C in assay buffer (20 mM Tris pH 7.5, 10 mM MgCl2, 2 mM DTT, 10 mM NaCl, 200 mM ATP). Reactions were quenched, proteins were resolved by SDS-PAGE, subjected to in-gel digestion, and phosphate incorporation was assessed by LC-MS and LC-MS/MS.
2.3 Mass spectrometry
Mass spectrometry was performed using an Agilent 1100 series HPLC (Agilent Technologies) coupled to an LCQ Classic ion trap mass spectrometer (ThermoElectron). Peptides were loaded on to a Paradigm Platinum Peptide Nanotrap (Microm) then separated on a reverse-phase capillary column (10 cm × 75 μm, Jupiter Proteo C12, Phenomenex) with a linear gradient from 2%-40% acetonitrile in 40 min. One full mass scan was acquired (300-2000 Da) and then the four most intense peaks were selected for MS/MS analysis. The dynamic exclusion limit was set to exclude a given m/z after it had been sequenced 2x during a 90 sec interval. Mass spectra were analyzed using TurboSequest configured with the following parameters: a peptide mass tolerance of 2.5 Da (avg), a fragment ion mass tolerance of 1.0 Da (avg), differential modification on S/T/Y +80 Da, and allowance of two incomplete cleavages. All MS/MS peak assignments were manually validated. The relative abundance of the modified forms for each peptide was determined using the ICIS peak detection method (Xcalibur) for the indicated extract ions. The percentages were calculated based on the sum of integrated peak areas for all detected modified forms of the peptide.
2.4. cDNA constructs
To construct mutants CaVβ2bS152A/E and CaVβ2bT164A/D mutagenic primers were designed that contained an internal HindIII restriction site. CaVβ2bS152A/E and CaVβ2bT164A/D were all amplified in a two-phase manner, using a SacII-XhoI cDNA fragment of CaVβ2b or CaV1.2 in pBluescript SK+. Phase I generated the 5′ and the 3′ arms using the WT DNA template. PCR products were gel purified. Phase II combined the 5′ and 3′ arms to serve as both initial primers and template. PCR products were precipitated, washed, dried, and resuspended. PCR products and vector were cut with Sac II and Xho I and isolated by ethanol precipitation. The digested vector was treated with calf alkaline phosphatase (CIP, NEB). Digested DNA was run out on a 1.25% agarose gel. Fragment bands were excised and purified either by Spin-X column (Fisher Scientific) or QIAQuick (QIAGEN). Fragments were ligated (Fast-Link, Epicentre). A negative control of vector and no insert was run at the same time. Ligation mixtures were then transformed into competent DH5 cells and samples were plated onto LB plates overnight. DNA was extracted from mutant colonies by basic SDS/sodium acetate method. Samples were screened by cutting with restriction enzymes whose recognition sites were silently built into the mutation primer. Positive samples were further purified (Quantum miniprep kit, Bio-Rad Laboratories) and sequenced (BigDye, ABI). When the sequences were confirmed to be correct, DNA from the original mini-prep was retransformed, amplified, and extracted (QUANTUM maxiprep kit, Bio-Rad Laboratories). Each mutant DNA in pBluescript SK+ (Stratagene) and the full-length WT CaVα1.2 subunit in pCDNA3 (Stratagene) were digested with Sac II and Xho I, ligated, and subcloned. Samples were sequenced again to confirm the sequence of the final construct.
2.5. Cell culture
TsA-201 cells were grown to 80% confluence and transfected with an equimolar ratio of cDNA encoding CaV1.2 full-length (FL), CaVβ2b or mutated CaVβ2b (CaVβ2bS152A/E and CaVβ2bT164A/D, CaVα2δ1, and CD8 as a cell surface marker (EBO-pCD-Leu2; American Type Culture Collection) using Fugene (Roche). 15–24 h after transfection, cells were suspended, plated at low density in 35-mm dishes, and incubated at 37°C in 10% CO2 for at least 17 h before recording using the whole-cell configuration of the patch clamp technique. Transiently transected cells were visualized with latex beads conjugated to an anti-CD8 antibody (Invitrogen).
2.6. Electrophysiology
Patch pipettes (2.5–3.5 MΩ) were pulled from micropipette glass (VWR Scientific) and fire-polished. Currents were recorded with an Axopatch 200B amplifier (MDS Analytical Technologies) and sampled at 5 kHz after anti-alias filtering at 2 kHz. Data acquisition and command potentials were controlled by Pulse (Pulse 8.50; HEKA), and data were stored for off-line analysis. Voltage protocols were delivered at 10s intervals unless otherwise noted, and leak and capacitive transients were subtracted using a P/4 protocol. Approximately 80% of series resistance was compensated with the voltage clamp circuitry.
For whole-cell voltage clamp recordings of CaV1.2 current in tsA-201 cells with Ba2+ or Ca2+ as a charge carrier (ICaV1.2(Ba/Ca)), the extracellular bath solution contained (in mM): 10 BaCl2 (or 1.8 mM CaCl2), 140 Tris, 2 MgCl2, and 10 D-glucose, titrated to pH 7.3 with MeSO4. The intracellular solution contained (in mM): 130 CsCl, 10 HEPES, 4 MgATP, 1 MgCl2, and 10 EGTA titrated to pH 7.3 with CsOH [32]. When Na+ was used as charge carrier (ICaV1.2(Na)), the extracellular solution contained (in mM): 150 NaCl, 10 HEPES, 0.2 MgCl2, 0.25 µM EDTA, and 10 D-glucose titrated to pH 7.3 with MeSO4. The intracellular solution contained (in mM): 150 CsOH, 110 glutamate, 20 HCl, 10 HEPES, 5 TrisATP, 4.3 MgCl2, and 10 EGTA titrated to pH 7.6 with CsOH [32].
2.7. Data analysis
Voltage-clamp data were compiled and analyzed using Igor (IGOR Pro version 5.0, Wavemetrics Inc.) and Excel (Excel 97, Microsoft). For measurement of CaV1.2 I-V relationship and voltage dependence of activation, peak step currents were measured from a 20 ms depolarization to potentials between −50 and 80 mV, while tail currents were measured during the subsequent repolarization to −40 mV. For the measurement of the voltage dependence of inactivation, 4-s depolarizations to potentials from −80 to 30 mV were applied to inactivate a fraction of Cav1.2 channels. A standard test pulse of 30 ms to 30 mV was applied, and peak tail currents were measured during repolarization to −40 mV immediately following the test pulse. Activation and inactivation data were fit to a Boltzmann function (Prism 5.0d, GraphPad Software Inc.). To normalize for variation in transfection efficiency currents were normalized to the gating charge (Qon)[33]. For CaV1.2 inactivation, currents were elicited by test pulses between −80 mV to 30 mV of 1000 ms duration. To quantify inactivation, peak currents elicited by 1000 ms depolarizations to 0 mV were normalized to 1.0, and the fraction of peak current remaining at the end of the voltage pulse (r1000) was measured. Pulses were applied every 30 s.
All data are presented as mean ± SEM. The statistical significance of differences between the various experimental groups was evaluated using the Student’s t test or one-way ANOVA, followed by the Newman-Keuls post-test; p-values are presented in the text.
Results
3.1. Sites of phosphorylation of CaVβ1a subunits
CaV1.1 channels in partially purified preparations of skeletal muscle transverse tubule membranes were labeled with [3H]isradipine, solubilized with digitonin, and purified by chromatography on wheat germ agglutinin-Sepharose and DEAE-Sepharose [30, 31, 34]. The purified protein was concentrated by re-chromatography on wheat germ agglutinin-Sepharose, and the subunits were separated by SDS-PAGE (Fig. 1A). The band at approximately 55 kDa containing the CaVβ1a subunit was excised, extracted, and prepared for mass spectrometry as described under Experimental Procedures (Fig. 1A). Analysis of the mass spectra revealed phosphorylation of Ser193 in the peptide Ser186-Arg202, which has a molecular mass of 820.4 without phosphorylation and 860.4 after phosphorylation (Fig. 2). In addition, we observed phosphorylation of Thr205 in the peptide Arg203-Lys240 (Fig. 3). Both of these sites are located in the HOOK domain, which connects the SH3 domain and the guanylate kinase domain (Fig. 1B). The functional role of the HOOK domain is unknown.
Figure 1. Sites of phosphorylation of CaVβ1a in CaV1.1 channels in skeletal muscle.

A. CaV1.1 channels were isolated from rabbit skeletal muscle as described in Experimental Procedures. Subunits were separated by SDS-PAGE and stained with Commassie blue. The protein band containing CaVβ1a was excised as illustrated and used for analysis by mass spectrometry. B. Alignment of the amino acid sequences of the CaVβ subunits with secondary structure motifs indicated. Stars, Ser193 and Thr205.
Figure 2. Identification of phosphorylated Ser193 by mass spectrometry.

CaVβ1a was isolated and analyzed as described in Experimental Procedures. The MS/MS spectrum of the phosphorylated tryptic peptides of CaVβ1a (Parent MH2+ 860.4) is illustrated. For clarity, only the relevant portion of the spectrum is shown.
Figure 3. Identification of phosphorylated Thr205 by mass spectrometry.

CaVβ1a was isolated and analyzed as described in Experimental Procedures. The MS/MS spectrum of the phosphorylated tryptic peptides of CaVβ1a (Parent MH3+ 1385.3) is illustrated. For clarity, only the relevant portion of the spectrum is shown.
Ser193 is in the amino acid sequence -SSLGD/E-, which is conserved in all four CaVβ subunits (Fig. 1B). The analogous sequence in CaVβ2b, the most abundant isoform in mammalian heart [35], contains the potential phosphorylation target Ser152. The consensus sequence SXXD/E is a potential site for phosphorylation by casein kinase II (www.phosphosite.org). However, the level of phosphorylation of this site in vivo was low, and casein kinase II treatment did not increase phosphorylation of this peptide in purified CaV1.1 channels in vitro (Fig. 4). This site may be phosphorylated at a low level by another kinase in vivo, or it may only be phosphorylated by casein kinase II under specific physiological conditions in vivo.
Figure 4. Phosphorylation of Ser193 by casein kinase II.

Purified CaV1.1 channel was phosphorylated by purified casein kinase II as described in Experimental Procedures. The CaVβ1a subunit was isolated by SDS-PAGE and analyzed by MS/MS. Integrated intensities of the peptide peaks representing unphosphoryated CaVβ1a (0P) and phosphorylated CaVβ1a (1P) are illustrated.
Thr205 is in the amino acid sequence -RRTP-, which is a consensus for phosphorylation by PKA (Fig. 5). The Hook domain of CaVβ1 was previously shown to be phosphorylated by PKA at Thr205 in vitro in purified preparations of skeletal muscle calcium channels [18], and bioinformatic analysis suggests that CaVβ1T205 is within a strong consensus sequence for PKA/CaMKII/PKC phosphorylation [18, 27]. This site is conserved in CaVβ1, CaVβ2, and CaVβ4, although the consensus is less optimal for phosphorylation in CaVβ4 because one of the two Arg residues is replaced by Phe (Fig. 1B). Incubation of purified CaV1.1 channels with PKA in vitro substantially increased the phosphorylation of Thr205 (Fig. 5), consistent with a significant physiological role of phosphorylation of this site. This sequence is conserved as -RKST- in CaVβ2b with Thr164 as the phosphorylated residue.
Figure 5. Phosphorylation of Thr205 by PKA.

Purified CaV1.1 channel was phosphorylated by purified PKA as described in Experimental Procedures. The CaVβ1a subunit was isolated by SDS-PAGE and analyzed by MS/MS. Integrated intensities of the peptide peaks representing unphosphoryated CaVβ1a (0P) and phosphorylated CaVβ1a (1P) are illustrated.
3.2. Modulation of CaV1.2 channel activity by mutation of Ser152 in CaVβ2b
CaV1.2 channels have multiple sites of phosphorylation on their pore-forming α1 subunits, which complicates interpretation of experiments that manipulate phosphorylation of CaVβ subunits by activation of protein kinases. Moreover, the kinase(s) that phosphorylate Ser193/Ser152 in vivo are unknown. Therefore, to probe potential functional effects for phosphorylation of this site in CaV1.2 channel regulation, we created phosphomimetic and phosphoinhibitory mutants by substitution of Glu and Ala, respectively, for Ser152. We expressed CaV1.2 channels with the CaVβ2b mutants CaVβ2bS152A and CaVβ2bS152E in human embryonic kidney tsA-201 cells and processed them for electrophysiological analysis. The phosphoinhibitory mutation, CaVβ2bS152A, increased CaV1.2 channel current amplitude, whereas phosphomimetic mutation, CaVβ2bS152E, decreased CaV1.2 current amplitude relative to CaVβ2bWT (Fig. 7A). The CaV1.2 current amplitudes were −6.49 ± 0.9 pA/fC (n = 8) and −2.95 ± 0.88 pA/fC (n = 6) (P < 0.05), for CaVβ2bS152A and CaVβ2bS152E, respectively. The phosphomimetic mutation CaVβ2bS152E also shifted the I/V relationship and the voltage dependence of activation and inactivation to more positive membrane potentials, whereas the phosphoinhibitory mutation CaVβ2bS152A had the opposite effects (Fig. 6A-C). The activation V1/2 values were −11.1 ± 2.2 mV (n = 8) and 7.5 ± 5.2 mV (n = 6) (P < 0.05), for CaVβ2bS152A and CaVβ2bS152E, respectively, whereas the activation V1/2 for CaVβ2bWT was intermediate at −1.0 ± 2.7 mV (n = 12). Similar results were observed with these phosphomimetic mutations inserted into CaVβ1b. Overall, these results suggest that introduction of negative charge at this novel phosphorylation site in the HOOK domain of CaVβ subunits acts as a gating modifier to reduce opening probability of CaV1.2 channels and to impede the voltage-induced activation and inactivation gating of CaV1.2 channels.
Figure 7. CaVβ2b Hook-domain phosphorylation at Thr164 does not alter current amplitude, voltage dependence of activation or inactivation.

A. Effect of “phosphorylation mimetics” of CaVβ2bT164 on CaV1.2 I-V relationship. Ca2+ was the charge carrier and in order to normalize for CaV1.2 channel expression we divided the current amplitude by gating current (Qon)[32]. B. CaVβ2bT164D and CaVβ2bT164A did not alter the voltage dependence ofactivation of CaV1.2 channels, or C. voltage dependence of inactivation of CaV1.2 channels. CaVβ2bWT (n = 8, 8, 4), CaVβ2bT164A (n = 6, 6, 4) and CaVβ2bT164D (n = 9, 9, 6) for panel A, B and C, respectively. Activation (Act.) and inactivation (Inact.).
Figure 6. CaVβ2b Hook-domain phosphorylation at Ser152 attenuates CaV1.2 current and shifts to the right the voltage dependence of activation of CaV1.2 channels.
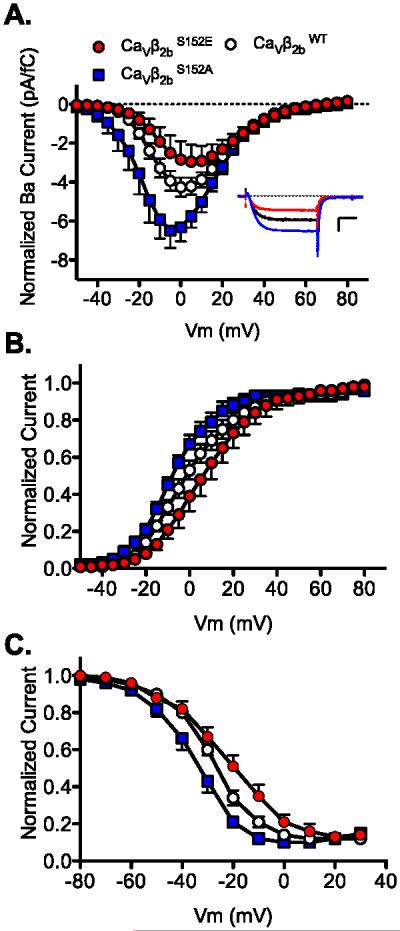
A. Effect of “phosphorylation mimetics” of a novel CK2 phosphorylation site in CaVβ2bS152 on CaV1.2 channel I-V relationship. Ba2+ was the charge carrier and in order to normalize for CaV1.2 channel expression we divided the current amplitude by gating current (Qon)[32]. Inset, representative normalized typical current traces of CaVβ2bS152A, CaVβ2bS152E, and CaVβ2bWT taken at the peak of the I-V relationship (calibration bar: 5 ms and 2.5 pA/fC. The dotted line represents zero current level). B. CaVβ2bS152A shifts to the left the voltage dependence of activation of Ca 1.2 channels while CaVβ2bS152E shifts to the right the voltage dependence of activation of Ca 1.2 channels relative to CaVβ2bWT. C. CaVβ2bS152A shifts to the left the voltage dependence of inactivation of CaV1.2 channels while CaVβ2bS152E shifted to the right the voltage dependence of inactivation of CaV1.2 channels relative to CaVβ2bWT. Activation and inactivation parameters were: CaV1.2 FLWT: V1/2 = −1.0 ± 2.7 mV, k = −12.1 ± 1.0, n = 12 and V1/2 = −28.0 ± 1.0 mV, k = 8.7 ± 0.6, n = 11; CaVβ2bS152A: V1/2 = − 11.1 ± 2.2 mV, k = −11.5 ± 2.2, n = 8 and V1/2 = −35.0 ± 2.3 mV, k = 7.8 ± 0.5, n = 8; and CaVβ2bS152E: V1/2 = 7.5 ± 5.2 mV, k = − 11.7 ± 1.8, n = 6 and V1/2 = −22.7 ± 3.0 mV, k = 12.3 ± 0.8, n = 6 for activation and inactivation parameters, respectively. There is a significant difference in V1/2 of activation (≈18 mV, P < 0.05) and V1/2 SS-inactivation (≈12 mV, P < 0.01) between CaVβ2bS152A and CaVβ2bS152A.
3.3. Modulation of CaV1.2 channel activity by mutation of CaVβ2b Thr164
To probe the role of phosphorylation of Thr164 in CaV1.2 channel regulation, we expressed CaV1.2 channels with the phosphoinhibitory and phosphomimetic mutations CaVβ2bT164A and CaVβ2bT164D in tsA-201 cells and performed electrophysiological analysis in the presence of the physiological charge carrier Ca2+ (1.8 mM). These mutations did not have strong effects on the I/V relationship or the voltage dependence of activation and inactivation (Fig. 7). However, we observed enhanced rate and extent of inactivation of CaV1.2 channels with WT and CaVβ2bT164D subunits compared to CaVβ2bT164A subunits (Fig. 8A and B). Analysis of two-exponential fits of these results showed that these effects were primarily on the second time constant for inactivation (Table 1). The results for WT CaVβ were similar to T164D, suggesting that this site is at least partially phosphorylated when Ca2+ currents are measured in normal physiological medium. We also engineered this mutation in CaVβ2a to test if this effect would be present with a CaVβ subunit that confers decreased inactivation to CaV1.2 channels because of its membrane tethering by N-terminal palmitoylation [36]. CaV1.2 channels with CaVβ2aT164A or CaVβ2aT164D mutants had similar inactivation properties (Fig. 8C and D; Table 1). These results suggest that negative charge at CaVβ2bT164 enhances the transition of CaV1.2 channels from open to inactivated state by accelerating the second rate constant governing this inactivation transition and that N-terminal palmitoylation of the CaVβ2a subunit [36] overrides this effect.
Figure 8. CaVβ2b Hook-domain phosphorylation at Thr164 enhanced Ca2+-dependent inactivation (CDI) of CaV1.2 channels.

A. “Phosphorylation mimetics” CaVβ2bT164D enhances CaV1.2 channel inactivation. Currents were elicited by 1000 ms steps to 0 mV from a holding potential of −80 mV with Ca2+ as charge carrier. Average traces are represented. The dotted line in panels A, C, and E represent zero current level. CaVβ2bT164D: n = 12 and CaVβ2bT164A: n = 10. B. Bar graph of mean r1000 values measured with CaVβ2bT164D and CaVβ2bT164A were significantly different (* P < 0.01). C. Effects of CaVβ2aT164D and CaVβ2aT164A on CaV1.2 channel inactivation. CaVβ2aT164D: n = 4; CaVβ2aT164A: n = 4. D. Bar graph of mean r1000 values measured with CaVβ2aT164D and CaVβ2aT164A were not significantly different (NS). E. Effect of “phosphorylation mimetics” of CaVβ2aT164 on CaV1.2 channel inactivation with Na+ as charge carrier. CaVβ2bT164D: n = 11; CaVβ2aT164A: n = 8. F. Bar graph of mean r1000 values measured with CaVβ2aT164A with Na+ as charge carrier were not significantly different.
Table 1.
| A1 | t1 (msec) | A2 | t2 (msec) | N | |||||
|---|---|---|---|---|---|---|---|---|---|
| CaVb2b | |||||||||
| WT | 0.53 | ±0.03 | 612 | ±54 | 0.45 | ±0.05 | 155 | ±37 | 13 |
| T164A | 0.34 | ±0.05 | 688 | ±77 | 0.29 | ±0.03 | 132 | ±24 | 10 |
| T164D | 0.56 | ±0.06 | 626 | ±155 | 0.37 | ±0.02 | 146 | ±34 | 12 |
| CaVb2a | |||||||||
| T164A | 0.27 | ±0.07 | 528 | ±331 | 0.27 | ±0.09 | 56 | ±15 | 4 |
| T164D | 0.21 | ±0.04 | 834 | ±89 | 0.33 | ±0.04 | 76 | ±5 | 4 |
Both voltage-dependent inactivation and Ca2+-dependent inactivation contribute substantially to the inactivation of CaV1.2 channels when Ca2+ is the charge carrier. In order to assess the effects of the CaVβ2bT164 mutations on voltage-dependent inactivation of CaV1.2 channels specifically, we used Na+ as charge carrier [32]. We did not observe any significant difference in voltage-dependent inactivation between CaVβ2bT164D and CaVβ2bT164A (Fig. 8E and F). These results suggest that negative charge at the position of CaVβ2bT164 enhances the Ca2+-dependent transition of CaV1.2 channels to the inactivated state with little effect on voltage-dependent inactivation. The results for WT CaVβ2b revealed slightly faster inactivation (Fig. 8E and F), as if the hydroxyl group of the native Ser at this position makes an interaction that slightly enhances the rate of voltage-dependent inactivation, and this interaction is prevented in both mutants.
4. Discussion
4.1. nctional effects of phosphorylation sites in the C-terminal of CaVβ subunits
Previous studies have identified multiple sites of phosphorylation in the C-terminal domain of CaVβ subunits by protein kinases in vitro [18, 27-29]. PKA phosphorylates three sites in the C-terminal domain of CaVβ2b subunits, and phosphorylation of these sites was reported to regulate the function of CaV1.2 channels expressed in nonmuscle cells [27]. Phosphatidylinositol-3 kinase phosphorylates a site in the C-terminal domain of the CaVβ2 subunit and regulates trafficking of the calcium channel complex to the plasma membrane [28]. Protein kinase G also phosphorylates a site in the C-terminal domain of the CaVβ2b subunit and inhibits the activity of CaV1.2 channels expressed in nonmuscle cells [10]. Although these studies suggest essential functional roles for the C-terminal domain of CaVβ2 subunits, deletion of the C-terminal domain including all three proposed sites of regulatory phosphorylation has no effect in mice [6]. The resulting C-terminal knockout mice are viable, fertile, and have no apparent physiological deficits [6]. Moreover, stimulation of the L-type calcium current in ventricular myocytes from these mice is normal [6]. Thus, these C-terminal phosphorylation sites on CaVβ2 subunits do not have essential functional roles that have been demonstrated in vivo. It is not known whether other CaVβ subunits can effectively compensate for loss of regulation by phosphorylation at these sites in the CaVβ2 subunit, but CaVβ subunits do compensate for each other when the complete genes are deleted [37]. Further work is required to determine whether the functional effects of phosphorylation of the C-terminal domain of CaVβ2 subunits observed in vitro in transfected nonmuscle cells are also important in vivo.
4.2. Phosphorylation sites in the Hook domain of CaVβ2b subunits
Unexpectedly, our mass spectrometry analysis did not detect phosphorylation of the C-terminal domain of the CaVβ1a subunit, but we found two previously unidentified sites of in-vivo phosphorylation in the Hook domain: Thr205 and Ser193. These sites are located far from the site of interaction of CaVβ subunits α1 subunits (Fig. 9, AID) and far from the previously identified sites of phosphorylation in the C-terminal region. Thr205 was identified as a PKA phosphorylation site by in vitro phosphorylation with [γ32P]ATP in early biochemical studies of purified CaV1.1 channels [18], and we confirmed those results with mass spectrometry in this work. Ser193 was not previously identified as a site of protein phosphorylation of CaVβ1a in biochemical studies, and the kinase that is responsible for its phosphorylation in vivo remains unknown.
Figure 9. Structural model of the CaVβ subunit.
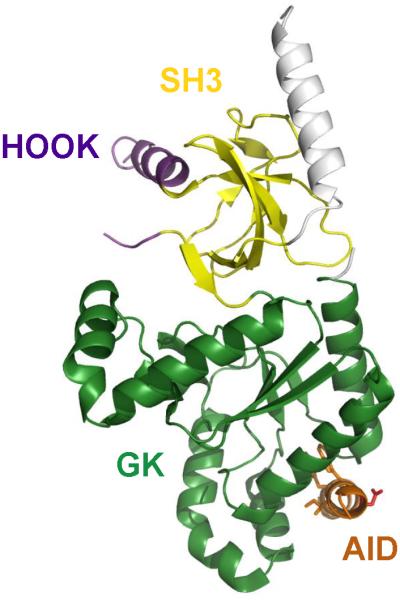
The SH3 domain (yellow), HOOK domain (purple), Alpha Interaction Domain (AID, red), and Guanylate Kinase domain (GK, green) are illustrated in the indicated colors.
Functional studies of CaV1.1 channels are made difficult by poor cell-surface expression in nonmuscle cells. Therefore, we analyzed the potential functional effects of phosphorylation of the homologous Ser152 and Thr164 residues through studies of mutants of the CaVβ2b subunit co-expressed with CaV1.2 channels in human embryonic kidney tsA-201 cells. The functional effects of phosphomimetic and phosphoinhibitory mutations at these sites suggest complementary regulatory roles. Addition of a negative charge at Ser152 decreases peak CaV1.2 channel currents and shifts both activation and inactivation to more positive membrane potentials. Together, these effects would significantly decrease channel activity. Because co-expression of CaVβ2b subunits causes a negative shift in the voltage dependence of channel activation [38], addition of negative charge at this position by protein phosphorylation would oppose this functional effect of the CaVβ2b subunit. In contrast, addition of negative charge at the position of Thr164 in CaVβ2b subunits has no effect on the voltage dependence of activation or inactivation, but it significantly increases the rate of calcium-dependent inactivation. These results suggest possible interactions between the Hook domain in the CaVβ2b subunit and the proximal C-terminal domain of the CaVα1 subunit where calcium-dependent inactivation is mediated by interaction of calcium/calmodulin with an IQ motif [15].
4.3. Possible physiological significance of phosphorylation of the Hook domain
Our results revealing functional effects of phosphomimetic mutations in the Hook domain are in agreement with previous work showing that the Hook domain of CaVβ can regulate CaV channel inactivation [39]. Deletion of the Hook domain of CaVβ2a enhanced CaV2.2 channel inactivation suggesting that the Hook domain normally interacts with CaV2.2 α-subunit to impede CaV2.2 channel inactivation [39]. The CaVβ Hook domain was also shown to regulate the binding affinity of Gβγ to CaVβ subunits to regulate CaV2.2 channel activity [40]. It is possible that phosphorylation sites in the Hook domain could regulate the binding affinity of Gβγ for CaVβ subunits and thereby modulate their effects on CaV2.2 channel activity because the interaction sites for Gβγ and CaVβ-subunits are located close to each other in the intracellular linker connecting domains I and II of CaVα1 subunits [41-45].
Determining the significance of phosphorylation of the Hook domain of CaVβ-subunits for regulation of basal and PKA-stimulated activity of CaV1.2 channels is an important aim for future experiments, but will require a detailed analysis of the complex signaling network that controls the activity of this channel. The basal activity of CaV1.2 channels expressed as an autoinhibitory signaling complex with bound AKAP and PKA in nonmuscle cells is much reduced by mutations that prevent phosphorylation of Ser1700 and Thr1704, which are located at the interface between the distal and proximal C-terminal domains of the CaVα1 subunit [23, 24]. Ser1700 is a substrate for phosphorylation by PKA and CaMKII, whereas Thr1704 is a substrate for casein kinase II [23, 24]. If phosphorylation of Ser152 in CaVβ2b inhibits CaV1.2 channel activity, as implied by our results, this effect would oppose up-regulation of basal channel activity by phosphorylation at Ser1700 and Thr1704 in the CaVα1 subunit.
β-adrenergic stimulation of the heart in the fight-or-flight response leads to both increased contractility, which is caused by increased peak L-type calcium currents in atrial and ventricular myocytes, and to increased beating rate, which is caused jointly by activation of hyperpolarization- and cyclic nucleotide-gated channels and CaV1.3 channels in the sino-atrial node [46, 47]. PKA stimulation of CaV1.2 channels expressed as an autoinhibitory signaling complex in nonmuscle cells is blocked by mutations that prevent phosphorylation of Ser1700 in the CaVα1 subunit [23], and mutation of Ser1700 and Thr1704 in mice greatly reduces the response to β-adrenergic stimulation in ventricular myocytes [25, 26]. If phosphorylation of Thr164 in the CaVβ2b subunit has no effect on peak current, as implied by our results, this site of phosphorylation would not contribute directly to the up-regulation of CaV1.2 channel activity by PKA. However, increased beating rate in response to β-adrenergic stimulation of the heart requires shortening of the ventricular action potential. Enhanced activation of KV channels by PKA phosphorylation is a well-documented mechanism contributing to shortening of the ventricular action potential during β-adrenergic stimulation of the heart [48]. However, phosphorylation of Thr164 in the CaVβ2b subunit would increase the rate of calcium-dependent inactivation, shorten the L-type calcium current, and thereby contribute to shortening the plateau of the ventricular action potential. Knock-in mutations in mice will be require to rigorously test these potential physiological effects of phosphorylation of the Hook domain of CaVβ2b subunits.
Highlights.
Ca2+ channel β subunits are phosphorylated on two sites in the Hook domain in vivo
Ser152 in CaVβ2b has a casein kinase II consensus sequence; Thr 164 is a PKA site
Phosphomimetic mutation S152E in CaVβ2b decreased peak current and shifted activation
Phosphomimetic mutation T164D in CaVβ2b increased Ca2+-dependent inactivation
Phosphorylation of sites in the Hook domain may regulate CaV1.2 channels in vivo
Acknowledgements
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health under award number R01HL085372. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- [1].Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–52. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- [3].Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–74. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- [4].Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation by β-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol. 1986;18:691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- [5].Trautwein W, Cavalie A, Flockerzi V, Hofmann F, Pelzer D. Modulation of calcium channel function by phosphorylation in guinea pig ventricular cells and phospholipid bilayer membranes. Circ Res. 1987;61:I17–23. [PubMed] [Google Scholar]
- [6].Blaich A, Welling A, Fischer S, Wegener JW, Kostner K, Hofmann F, et al. Facilitation of murine cardiac L-type CaV1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A. 2010;107:10285–9. doi: 10.1073/pnas.0914287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen X, Piacentino V, 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–24. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- [8].Chen X, Zhang X, Harris DM, Piacentino V, 3rd, Berretta RM, Margulies KB, et al. Reduced effects of BAY K 8644 on L-type Ca2+ current in failing human cardiac myocytes are related to abnormal adrenergic regulation. Am J Physiol Heart Circ Physiol. 2008;294:H2257–67. doi: 10.1152/ajpheart.01335.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, et al. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–76. doi: 10.1161/01.cir.98.10.969. [DOI] [PubMed] [Google Scholar]
- [10].Yang L, Katchman A, Morrow JP, Doshi D, Marx SO. Cardiac L-type calcium channel (CaV1.2) associates with γ subunits. FASEB J. 2011;25:928–36. doi: 10.1096/fj.10-172353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chu PJ, Robertson HM, Best PM. Calcium channel γ subunits provide insights into the evolution of this gene family. Gene. 2001;280:37–48. doi: 10.1016/s0378-1119(01)00738-7. [DOI] [PubMed] [Google Scholar]
- [12].Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–4. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the α 1 subunit of the cardiac L-type calcium channel by adenosine 3',5'-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35:10392–402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- [14].Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ - dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–58. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- [15].Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- [16].Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. β-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–8. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG, 2nd, Bigelow DJ, et al. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102:5274–9. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].De Jongh KS, Merrick DK, Catterall WA. Subunits of purified calcium channels: a 212-kDa form of α1 and partial amino acid sequence of a phosphorylation site of an independent β subunit. Proc Natl Acad Sci U S A. 1989;86:8585–9. doi: 10.1073/pnas.86.21.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Jongh KS, Warner C, Colvin AA, Catterall WA. Characterization of the two size forms of the α1 subunit of skeletal muscle L-type calcium channels. Proc Natl Acad Sci U S A. 1991;88:10778–82. doi: 10.1073/pnas.88.23.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hulme JT, Ahn M, Hauschka SD, Scheuer T, Catterall WA. A novel leucine zipper targets AKAP15 and cyclic AMP-dependent protein kinase to the C terminus of the skeletal muscle Ca2+ channel and modulates its function. J Biol Chem. 2002;277:4079–87. doi: 10.1074/jbc.M109814200. [DOI] [PubMed] [Google Scholar]
- [22].Brunet S, Scheuer T, Catterall WA. Increased intracellular magnesium attenuates β-adrenergic stimulation of the cardiac CaV1.2 channel. J Gen Physiol. 2013;141:85–94. doi: 10.1085/jgp.201210864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Emrick MA, Sadilek M, Konoki K, Catterall WA. β-adrenergic-regulated phosphorylation of the skeletal muscle CaV1.1 channel in the fight-or-flight response. Proc Natl Acad Sci U S A. 2010;107:18712–7. doi: 10.1073/pnas.1012384107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fu Y, Westenbroek RE, Scheuer T, Catterall WA. Basal and β-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc Natl Acad Sci U S A. 2014;111:16598–603. doi: 10.1073/pnas.1419129111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fu Y, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. Proc Natl Acad Sci U S A. 2013;110:19621–6. doi: 10.1073/pnas.1319421110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gerhardstein BL, Puri TS, Chien AJ, Hosey MM. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the β2 subunit of L-type voltage-dependent calcium channels. Biochem. 1999;38:10361–70. doi: 10.1021/bi990896o. [DOI] [PubMed] [Google Scholar]
- [28].Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F, et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci. 2004;7:939–46. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- [29].Yang L, Liu G, Zakharov SI, Bellinger AM, Mongillo M, Marx SO. Protein kinase G phosphorylates CaV1.2 α1C and β2 subunits. Circ Res. 2007;101:465–74. doi: 10.1161/CIRCRESAHA.107.156976. [DOI] [PubMed] [Google Scholar]
- [30].Florio V, Striessnig J, Catterall WA. Purification and reconstitution of skeletal muscle calcium channels. Methods Enzymol. 1992;207:529–46. doi: 10.1016/0076-6879(92)07037-o. [DOI] [PubMed] [Google Scholar]
- [31].Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochem. 1984;23:2113–8. doi: 10.1021/bi00305a001. [DOI] [PubMed] [Google Scholar]
- [32].Brunet S, Scheuer T, Catterall WA. Cooperative regulation of CaV1.2 channels by intracellular Mg2+, the proximal C-terminal EF-hand, and the distal C-terminal domain. J Gen Physiol. 2009;134:81–94. doi: 10.1085/jgp.200910209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bean BP, Rios E. Nonlinear charge movement in mammalian cardiac ventricular cells. Components from Na and Ca channel gating. J Gen Physiol. 1989;94:65–93. doi: 10.1085/jgp.94.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Takahashi M, Seagar MJ, Jones JF, Reber BF, Catterall WA. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc Natl Acad Sci U S A. 1987;84:5478–82. doi: 10.1073/pnas.84.15.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chu PJ, Larsen JK, Chen CC, Best PM. Distribution and relative expression levels of calcium channel β subunits within the chambers of the rat heart. J Mol Cell Cardiol. 2004;36:423–34. doi: 10.1016/j.yjmcc.2003.12.012. [DOI] [PubMed] [Google Scholar]
- [36].Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. J Biol Chem. 1996;271:26465–8. doi: 10.1074/jbc.271.43.26465. [DOI] [PubMed] [Google Scholar]
- [37].Burgess DL, Biddlecome GH, McDonough SI, Diaz ME, Zilinski CA, Bean BP, et al. β subunit reshuffling modifies N- and P/Q-type Ca2+ channel subunit compositions in lethargic mouse brain. Mol Cell Neurosci. 1999;13:293–311. doi: 10.1006/mcne.1999.0748. [DOI] [PubMed] [Google Scholar]
- [38].Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, et al. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J Biol Chem. 1992;267:1792–7. [PubMed] [Google Scholar]
- [39].Richards MW, Leroy J, Pratt WS, Dolphin AC. The HOOK-domain between the SH3 and the GK domains of CaVβ subunits contains key determinants controlling calcium channel inactivation. Channels (Austin) 2007;1:92–101. doi: 10.4161/chan.4145. [DOI] [PubMed] [Google Scholar]
- [40].Dresviannikov AV, Page KM, Leroy J, Pratt WS, Dolphin AC. Determinants of the voltage dependence of G protein modulation within calcium channel β subunits. Pflugers Arch. 2009;457:743–56. doi: 10.1007/s00424-008-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature. 2004;429:671–5. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc Natl Acad Sci U S A. 1997;94:1512–6. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–6. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- [44].Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- [45].Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, et al. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–80. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- [46].Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- [47].Mangoni ME, Couette B, Marger L, Bourinet E, Striessnig J, Nargeot J. Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol. 2006;90:38–63. doi: 10.1016/j.pbiomolbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- [48].Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]