

Abstract
Graft-versus-host disease (GVHD) is the major complication of allogeneic hematopoietic cell transplantation (alloHCT), a potentially curative therapy for hematologic diseases. It has long been thought that murine bone marrow (BM) derived T cells do not mediate severe GVHD because of their quantity and/or phenotype. During the course of experiments testing the impact of housing temperatures on GVHD, we discovered that this apparent resistance is a function of the relatively cool ambient housing temperature. Murine BM-derived T cells have the ability to mediate severe GVHD in mice housed at a thermoneutral temperature. Specifically, mice housed at IACUC mandated, cool standard temperatures (~22°C) are more resistant to developing GVHD than mice housed at thermoneutral temperatures (~30°C). We learned that the mechanism underlying this housing-dependent immunosuppression is associated with increased norepinephrine production and excessive signaling through β-adrenergic receptor (β-AR) signaling which is increased when mice are cold stressed. Treatment of mice housed at 22°C with a β2-adrenergic antagonist reverses the norepinephrine driven suppression of GVHD and yields similar disease to mice housed at 30°C. Conversely, administering a β2-adrenergic agonist decreases GVHD in mice housed at 30°C. In further mechanistic studies using β2-adrenergic receptor deficient (β2-AR−/−) mice, we found that it is host cell β2-AR signaling that is essential for decreasing GVHD. These data reveal how baseline levels of β-AR signaling can influence murine GVHD and point to the feasibility of manipulation of β2-AR signaling to ameliorate GVHD in the clinical setting.
Introduction
GVHD limits the full therapeutic potential of alloHCT for many patients (1). To better understand the immunobiology underlying GVHD pre-clinical mouse models are often employed (2). However, a well-recognized aspect of many mouse models is that they do not develop severe GVHD when only BM cells are used for transplantation (3). There are several plausible explanations that may explain this resistance including a smaller number of T cells in the murine BM graft (4) or to an increased proportion of suppressive populations of T cells (3, 5). To offset the quantitative and/or qualitative dearth of GVHD-capable murine BM-derived T cells, additional T cells (from spleen or lymph nodes) are added to the BM graft in order to generate GVHD (3, 6).
Work from murine physiologists has long established that mice housed at the relatively cool, currently mandated ambient temperature in research facilities (typically ~22°C) experience a mild, but chronic cold stress yet maintain normal body temperature (7-9). Our previous findings have shown that standard housing temperatures impair maturation of antigen presenting cells (APC) (10) and inhibit T cell-dependent immune responses in tumor-bearing mice, while enhancing the frequency of immunosuppressive cells (7). Our recent work demonstrated that cold stress-induced phenotypes in tumor-bearing mice could be completely reversed by using pharmacological blockade of adrenergic receptors (β-blockers) involved with norepinephrine-dependent signaling (11). Recently, these same drugs reveal an important role for β-AR signaling in modulation of immune activity (12-15)._ENREF_7 Although acute stress-induced β-AR signaling can benefit the immune system in a variety of ways helping to protect the host from infections (16-19), other research demonstrates that long term norepinephrine-driven β-AR signaling can inhibit T cell function and increase immunosuppression. Notably, previous reports show that β-AR signaling can decrease APC function (12, 13), influence CD4+ T helper cell function (20), prevent lymphocyte egress from lymph nodes (15) and increase the suppressive capacity of regulatory T cells (Treg) (21). GVHD is known to be both dependent upon immune effector activity (mediated by APC and T cells) and influenced by immunosuppressive activity (mediated by regulatory T cells and suppressive cytokines) (6, 22).
Based on these previous findings, we wondered whether the long-recognized inability of murine BM-derived T cells to mediate GVHD in many experimental models is actually a function of an unrecognized variable, housing temperature. We hypothesized that the cool standard housing temperature could be dampening anti-host immune activity through a mechanism related to cold stress. To test this we investigated whether the inability of murine BM-derived T cells to mediate GVHD could be related to increased β-AR signaling in mice housed at standard temperatures. Our findings indicate that standard housing temperature of laboratory mice and the subsequent increase of β-AR signaling in the host explain, in large part, the suppressed ability of murine BM-derived T cells to mediate GVHD.
Materials and Methods
Animals
Male and female 8 week old C57BL/6 (H-2b), 8 week old male and female BALB/c (H-2d) and 8 week old female 129/SvJ (H-2b) mice were obtained from the NCI and maintained under specific pathogen free conditions following guidelines established by Institutional Animal Use and Care Committee at Roswell Park Cancer Institute. β2-adrenergic receptor deficient (β2-AR−/−) mice were generously provide by David Farrar (UT Southwestern Medical Center). All mice were housed at either 22°C or 30°C in Precision Refrigerated Plant-Growth Incubators (Thermo Fischer Scientific) for 2 weeks prior to the experimental start date. Humidity within incubators was controlled using Top Fin® Air Pump AIR 1000 and Top Fin® airline tubing.
Acute GVHD models
MHC-mismatched transplants were performed using C57BL/6 and WT or β2-AR−/− BALB/c mice as donors and hosts. BALB/c mice received 792 cGy and C57BL/6 mice 965 cGy irradiation from a [137Cs] source 24 hours prior to transplant. Unless otherwise indicated, BALB/c mice received 7×106 total BM IV from C57BL/6 mice. MHC-matched transplants were performed using 129/SvJ as donor and C57BL/6 as hosts. C57BL/6 mice received 965 cGy from a [137Cs] source 24 h prior to transplant. Male C57BL/6 mice were then IV injected with 2×106 female 129/SvJ bone marrow plus 7.5×106 splenocytes and weight loss was tracked. For experiments with T cell depleted bone marrow (TCD-BM), anti-CD90.2 microbeads were used (Miltenyi Biotec). Syngeneic transplants were performed using C57BL/6. C57BL/6 mice received 965 cGy from a [137Cs] source 24 hours prior to transplant, and were given 7×106 total BM IV from C57BL/6 mice. Following transplant, mice were monitored regularly for weight loss and survival. Mice were considered moribund once weight loss reached 20% of their initial weight. For alloHCT involving purified T cells, 3×106 TCD-BM with or without 0.5×106 PanT splenic T cells sorted from C57BL/6 mice were injected into BALB/c hosts. Conversely, C57BL/6 hosts received 4×106 WT BALB/c TCD-BM with or without 3×106 WT or β-AR−/− PanT cells.
β-adrenergic agonist/antagonist treatments
Starting on day 1 post-transplant, mice were injected daily IP with 10 mg/kg body weight of propranolol (Pro), metoprolol (Met) or salbutamol (Sal), 1 mg/kg body weight of ICI 118,551 (ICI) or 200 µl PBS diluent as a control.
Histopathological analysis of GVHD target organs
Liver and large and small intestines were removed, formalin-fixed, sectioned, and stained with H&E. Intestine tissues were examined using a previously established semi-quantitative scoring system(23, 24). Blinded assessments were made for the presence of crypt epithelial cell apoptosis, crypt loss, surface colonocyte vacuolization, surface colonocyte attenuation, lamina propria inflammatory cell infiltrate, mucosal ulceration, and luminal sloughing of cellular debris, leukocyte infiltration of the lamina propria, and villous blunting. Liver samples were evaluated using the clinical GVHD score system as previously described (25). Assessment for the percentage of pathologic small bile ducts designated 0 as normal, 1 as less than 25%, 2 as 25~49%, 3 as 50~75%, and 4 as above 75%. Representative pictures were captured at 100x.
Liver preparation
Livers were processed through a 70μM filter and flow-through was passed through a second 70μM filter. The cell suspension was centrifuged and then resuspended and mixed well in 35% Percoll in RPMI. This gradient was centrifuged for 20 min at 2000rpm at 4°C. Hepatocytes were then removed and remaining cells were red cell lysed before flow cytometry staining.
Flow cytometry
Single cell suspensions were stained with antibodies to H-2Kd (SF1-1.1), H-2Kb (AF6-88.5.5.3), CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), CD11b (M1/70), CD11c (N418) and fixable Live/Dead Aqua (Invitrogen). Cells were stained for 10 min at room temperature in 200μL PBS and washed once with FACS buffer. All samples were run on either an LSRFortessa (BD Biosciences). All data were analyzed with FlowJo.
Mixed lymphocyte reaction
Splenocytes were harvested from WT C57BL/6 mice and WT or β2-AR−/− BALB/c mice. Splenocytes from all 3 types of mice were red cell lysed and either stained with 500μM CFSE (responders) or given 3000 cGy irradiation from a [137Cs] source (stimulators). 0.25×106 CFSE+ responders and 0.75×106 irradiated stimulators were plated into 96-well round bottom plates and incubated for 96h as previously described (26). CFSE dilution in CD4+ and CD8+ T cells was analyzed by flow cytometry.
Results
Mild cold stress inhibits GVHD development
To determine whether ambient temperature affected GVHD, we utilized a standard MHC-mismatched alloHCT model. We transplanted lethally irradiated BALB/c (H-2d) mice housed at either 22 or 30°C with total BM from C57BL/6 (H-2b) mice. Consistent with the literature (3, 27), following alloHCT of BM with no additional T cells obtained from the spleen or lymph node, mice housed at standard temperature (22°C) show no signs of severe or lethal GVHD (Fig 1A and B). This was evidenced by no weight loss (Fig 1A) or lethality (Fig 1B) at 22°C. In contrast, we observed significant weight loss and decreased survival in mice given the identical transplant, but housed at 30°C (Fig 1A-B). We also followed a commonly accepted protocol by transplanting T cell depleted BM (TCD-BM) plus purified splenic T cells (PanT). As expected, adding T cells in the BM graft induced more severe GVHD as evidenced by aggravated weight loss. In addition, we found that mice housed at 30°C showed acute and substantial lethality while those housed at 22°C showed no lethality despite weight loss (Fig 1C-D). Similar results were also obtained in an MHC-matched, minor histocompatibility antigen-mismatched alloHCT model using 129/SvJ (H-2b) donor-derived BM plus total splenocytes and C57LB/6 (H-2b) hosts (Fig 1E-F). These data suggest that GVHD is significantly decreased when mice experience chronic mild cold stress.
Figure 1. GVHD is exacerbated following alloHCT in mice maintained at 30°C.

(A-D) BALB/c mice were transplanted as described in Materials and Methods. Following transplant of total BM alone (A-B) or TCD-BM + PanT (C-D), weight loss and survival of BALB/c hosts was monitored. (E, F) C57BL/6 mice were lethally irradiated (965 cGy) and transplanted on day -1 with 2×106 129/SvJ BM + 7.5×106 splenocytes on day 0. Weight loss (C) and survival (D) were subsequently monitored. (A, C, E) Data presented as mean ± SEM (n = 3-10; **** p < 0.0001; Two-way ANOVA). (B, D, F) Data presented percent survival (n = 10; ## p < 0.01; Mantel-Cox test).
Norepinephrine levels are significantly increased in cold stressed mice
Norepinephrine, a β-adrenergic agonist produced in response to stress, is increased in cold stressed mice (28). To assess the activation of a stress response associated with mild cold stress we measured serum norepinephrine in mice housed at 22 or 30°C. We found that naïve mice housed at 22°C had significantly higher norepinephrine levels than their counterparts at 30°C (Fig 2). Norepinephrine levels were also elevated at 22°C compared to 30°C on day 13 following alloHCT (Fig 2). Thus, pre- and post-alloHCT mice housed at 22°C are experiencing a greater degree of stress compared to mice housed at 30°C.
Figure 2. Mild cold stress increases circulating norepinephrine pre- and post-alloHCT.
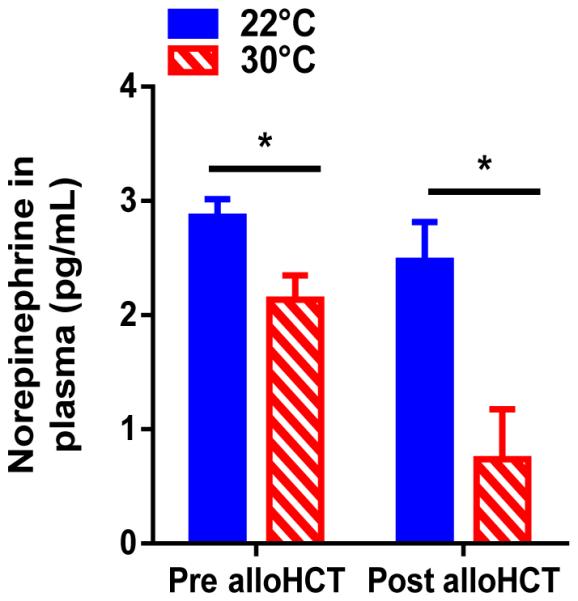
BALB/c mice were housed at 22°C or 30°C and transplanted as described in Materials and Methods. (A) Norepinephrine levels were measured in BALB/c mice prior to transplant and on day 13 following alloHCT. (A) Data presented as mean ± SEM (n = 5-8; *p < 0.05; Student’s t test).
Stress-induced adrenergic signaling suppresses GVHD mediated by allogeneic T cells
We next examined whether the resistance to GVHD observed in mice housed at 22°C was due to increased norepinephrine driven β-AR signaling. We blocked β-AR signaling with propranolol and housed mice at 22°C and 30°C. Following transplantation, GVHD was increased in propranolol-treated mice housed at 22°C (Fig 3A-B). At 30°C, there is minimal norepinephrine-driven stress, and propranolol treatment of these mice had no additional effect on the severity of GVHD above that seen by 30°C housing alone (Fig 3A-B). Thus, alleviating adrenergic stress by housing at 30°C or pharmacologically blocking β-AR signaling results in increased GVHD.
Figure 3. The adrenergic stress response reduces allogeneic T cell mediated GVHD.

(A-B) BALB/c mice were transplanted and treated as described in Materials and Methods. Mice were treated daily with propranolol or PBS while weight (A) and survival (B) were monitored. (C-D) C57BL/6 mice were transplanted with syngeneic BM as described in Materials and Methods. Host mice were treated daily with propranolol or PBS and weight loss (C) and survival (D) were monitored. (A, C, E) Data presented as mean ± SEM (n = 5-10; **** p < 0.0001; Two-way ANOVA). (B, D, F) Data presented as percent survival (n = 5-10; # p < 0.05, ## p < 0.01; Mantel-Cox test).
Previous work has demonstrated that either housing mice at 30°C (10) or treating with propranolol (29) could decrease body weight compared to controls. Thus, we were concerned that weight loss and possibly survival differences were not due to GVHD but to the specific experimental conditions. We therefore tested the likelihood that allogeneic graft-versus-host responses were responsible for changes in weight and survival. C57BL/6 mice underwent syngeneic transplant with BM from C57BL/6 donors. Neither housing temperature nor propranolol treatment affected weight loss or survival following syngeneic HCT (Fig 3C-D). Thus, an allogeneic response is required to increase GVHD severity in mice housed at 30°C or mice housed at 22°C and treated with propranolol. Next, to determine the importance of donor allogeneic T cells in the regulation of GVHD in mice with reduced β-AR signaling, we performed an alloHCT with a depletion of donor T cells from the BM graft. T cell depletion from the BM inoculum completely eliminated symptoms of GVHD in mice either housed at 30°C or treated with propranolol (Fig 3E-F). These data suggest that adrenergic stress down-regulates allogeneic T cell responses thereby diminishing GVHD severity.
Liver GVHD is significantly increased in mice with reduced adrenergic signaling
In addition to weight loss and survival for assessing GVHD, we performed histopathologic analyses to examine how β-AR signaling influence GVHD occurrence and severity in target organs. We harvested small intestine, large intestine and liver on day 54 following alloHCT for histopathologic analyses. Blinded histopathologic quantification using an established clinical GVHD scoring system (25) revealed significantly increased liver inflammation consistent with GVHD in the livers of mice housed at 30°C and/or treated with propranolol (Fig 4A-B). However, using a previously established semi-quantitative scoring system (23, 24), we found no major differences in GVHD score in the intestines (Fig 4C-D). These data suggest that blocking β-AR signaling significantly increases liver GVHD.
Figure 4. Alleviating adrenergic stress increases liver GVHD.

BALB/c mice were transplanted as described in Materials and Methods. 54 days following alloHCT livers (A, B) and small (C) and large (D) intestines were excised and fixed in formalin. GVHD was scored using a previously described semi-quantitative scoring system. (E-H) Livers were harvested day 5 post-alloHCT as described in Materials and Methods. Cells were counted by trypan blue exclusion using a hemocytometer and then analyzed by flow cytometry for the presence of CD3+CD4+ T cells (E), CD3+CD8+ T cells, CD11b+CD11c− (G), and CD11b−CD11c+ cell (H). Data are pooled from two individual experiments. (B-H) Data presented as mean ± SEM (n = 6; * p < 0.05;**p <0.01 Student’s t test).
We next characterized the inflammatory infiltrate in the livers of mice housed at 22°C or 30°C. We evaluated an early time point (day 5 post alloHCT) in which allogeneic donor T cells begin to enter the liver (30). Consistent with our pathologic GVHD data, both donor CD4+ and CD8+ T cells were substantially higher in the livers of mice housed at 30°C compared to those housed at 22°C (Fig 4E-F). Furthermore, we analyzed the infiltrate of donor-type APC and found that both CD11b+CD11c− and CD11b+CD11c+ cells were significantly increased in the livers of mice housed at 30°C versus 22°C (Fig 4G-H). Together, these cellular infiltrates are consistent with the increased liver pathology following alloHCT of mice housed at 30°C.
β2-but not β1-adrenergic signaling influences GVHD severity
As a pan β-antagonist, propranolol inhibits norepinephrine binding of both β1- and β2-adrenergic receptors we tested whether blockade of either class of receptor was sufficient to affect GVHD severity. Using metoprolol, a β1-AR antagonist, we found GVHD severity was unaffected in mice undergoing alloHCT housed at 22°C (Fig 5A-B). In contrast, treatment with ICI 118,551, a β2-AR antagonist, increased GVHD in mice housed at 22°C (Fig 5C-D). In line with our previous data using the pan β-AR antagonist propranolol, specifically blocking β1- or β2-AR signaling did not impact GVHD in mice housed at 30°C (Fig 5A-D). Together these data suggest that β2-AR signaling is responsible for decreased GVHD in mice housed at 22°C. Thus, we postulated that targeting β2-AR with a specific agonist would reduce GVHD in mice housed at 30°C. We used the β2-AR agonist salbutamol to determine if it would lessen the severity of GVHD. As expected, mice housed at 22°C, in which high norepinephrine levels already decrease GVHD, salbutamol treated mice did not develop GVHD following alloHCT (Fig 5E-F). However, salbutamol did decrease GVHD severity in mice housed at 30°C (Fig 5E-F). This demonstrates that specific modulation of β2-AR signaling can control GVHD.
Figure 5. β2- but not β1-adrenergic receptor signaling reduces GVHD.

BALB/c mice were transplanted and treated as described in Materials and Methods. Mice were treated daily with metoprolol, ICI 118,551, salbutamol or PBS while weight (A, C, D) and survival (B, D, F) were monitored. (A, C, E) Data presented as mean ± SEM (n = 5-10; **** p < 0.0001; Two-way ANOVA). (B, D, F) Data presented as percent survival (n = 5-10; # p < 0.05, ## p < 0.01; Mantel-Cox test).
β2-AR signaling decreases T cell proliferation intrinsically and extrinsically
While it appears that β2-AR signaling can reduce GVHD, the question remains as to what cell(s) are mediating this phenotype. To answer this question, we performed mixed lymphocyte reactions (MLR) to decipher if β2-AR signaling on the T cell or APC was responsible for decreasing GVHD. To determine the importance of T cell β2-AR signaling we cultured either WT or β2-AR−/− BALB/c T cell responders with irradiated WT C57BL/6 stimulators. We found that β2-AR−/− T cells proliferated more than WT T cells, suggesting that β2-AR signaling could directly decrease T cell proliferation in vitro (Fig 6A-B). We next asked whether β2-AR signaling could influence the ability of APC to stimulate T cell proliferation. In this system, we mixed WT C57BL/6 responders with irradiated WT or β2-AR−/− BALB/c stimulators. β2-AR−/− stimulators showed an increased ability to stimulate allogeneic T cell proliferation compared to WT controls (Fig 6 C-D). Therefore, the presence of β2-AR signaling on APC dampens their ability to stimulate T cell proliferation. Together these data indicate that in vitro T cell proliferation can be reduced by T cell (intrinsic) or APC (extrinsic) β2-AR signaling stimulated by endogenous norepinephrine present in the serum used for cell culture (11).
Figure 6. β2-AR signaling decreases T cell proliferation in mixed lymphocyte reactions.

(A-B) WT or β2-AR−/− BALB/c responders were CFSE stained and co-cultured with irradiated WT C57BL/6 stimulators for 96 hours. CFSE dilution in H-2Kd+CD4+ and H-2Kd+CD8+ T cells was analyzed. (C-D) WT C57BL/6 responders were CFSE stained and co-cultured with irradiated WT or β2-AR−/− BALB/c stimulators for 96 hours. CFSE dilution in H-2Kb+CD4+ and H-2Kb+CD8+ T cells was analyzed. Data are presented as mean ± SEM (*p < 0.05;**p <0.01 Student’s t test). Representative data from 1 of 2 individual experiments is presented.
GVHD is increased in the absence of host, but not donor T cell derived β2-AR signaling
Our in vitro data suggested that β2-AR signaling could affect T cell responses by directly decreasing T cell proliferation and decreasing the ability of APC to stimulate T cell proliferation. We sought to test whether these findings could be recapitulated in our in vivo GVHD model. To test the ability of T cell intrinsic β2-AR signaling, we isolated WT and β2-AR−/− BALB/c PanT cells and transplanted them with WT TCD-BM into WT C57BL/6 hosts. However, it appeared that this donor-host combination did not induce severe GVHD and the elimination of β2-AR signaling on donor T cells did not exacerbate GVHD as we expected (Fig 7A-B). Since host APC are essential for GVHD (22, 31, 32), we reasoned that host APC-derived β2-AR was responsible for controlling GVHD post alloHCT. To test this, we transplanted WT and β2-AR−/− BALB/c mice with total BM from WT C57BL/6 donors. We found that β2-AR−/− hosts had significantly more GVHD than WT controls (Fig 7C-D). These data demonstrate that host cell β2-AR signaling suppresses GVHD.
Figure 7. Host-, but not donor T cell-, derived β2-AR signaling is essential for controlling GVHD.

(A-B) WT C57BL/6 host mice were housed at 22°C and transplanted with BALB/c-derived TCD-BM + PanT as described in Materials and Methods. Weight loss (A) and survival (B) of C57BL/6 hosts were monitored. (C-D) WT or β2-AR−/− BALB/c host mice were housed at 22°C and transplanted with C57BL/6-derived total BM cells as described in Material and Methods. Weight loss (C) and survival (D) of WT and β2-AR−/− BALB/c hosts were monitored. (A, C) Data presented as mean ± SEM (n = 5-10; *p<0.05; Two-way ANOVA). (B, D) Data presented as percent survival (n = 9-10; # p < 0.05, Mantel-Cox test).
Discussion
This is the first demonstration that the severity of GVHD is a function of ambient temperature used to house mice, and we have learned that the mechanism underlying this effect is closely linked to the degree of β-AR signaling in host mice. We show that both a physiologic stress (i.e. mild cold stress) known to generate increased norepinephrine-induced adrenergic activity and pharmacologic stimulation of β2-AR signaling can significantly reduce GVHD severity. Importantly, the cold stress imposed by standard housing conditions accounts, in large part, for the long-standing observation that many mouse models appear to be resistant to GVHD following alloHCT with only BM-derived T cells. While previous studies have posited that the quantity or immunosuppressive nature of BM-derived T cells explains the lack of GVHD seen in many murine models (3-5), our data demonstrate that immune activity is highly dependent upon the room temperature used to house mice. Cold stress is countered by a norepinephrine driven adrenergic response designed to generate heat to aid in body temperature maintenance (33, 34). However since adrenergic receptors are expressed on myriad cell types, additional effects of norepinephrine signaling are possible. In our model, we have shown one of these additional effects to be the dampening of the immune response following alloHCT.
In particular, our data using β2-AR−/− mice demonstrate that this effect is dependent on host-derived β2-AR signaling. Host APC participate in the initiation of T cell activation during GVHD (22, 31, 32) and mild cold stress (10) and β-adrenergic stimulation (12, 13) suppress the ability of APC to activate T cells. These data suggest that host APC-derived β2-AR signaling may decrease the ability of APC to stimulate T cell mediated GVHD. These data are corroborated by the fact that β2-AR−/− stimulators showed an enhanced ability to stimulate T cell proliferation in an MLR. Work from our group and others has shown that anti-tumor activity of CD8+ T cells has been shown to be impaired by mild cold stress (7) while CD4+ T cell activity can be modulated by adrenergic signaling (14). Interestingly, it appears that in vitro β2-AR signaling can impair T cell proliferation (Fig 6), though in our in vivo model, GVHD was no different between mice receiving WT or β2-AR−/− PanT cells (Fig 7). This implies that direct donor T cell-derived β2-AR signaling is not as effective as host cell-derived β2-AR signaling in decreasing GVHD. However, this interpretation has to be taken precautiously in consideration of that the BALB/c to C57BL/6 alloHCT with high doses of purified T cells did not induce severe GVHD.
Our data suggest that β2-AR signaling may decrease donor T cell liver infiltration post alloHCT, which could account for less GVHD. Another plausible and likely interconnected mechanism is a deficit in T cell egress from the lymph nodes in mice with increased β2-AR signaling, as work from others has demonstrated that increased β2-AR signaling can inhibit T cell egress from the lymph node resulting in reduced autoimmunity (15). This inhibition of T cell egress could contribute to reduced symptoms of GVHD. Further, non-hematopoietic cells (i.e. hepatocytes) can respond to, and be protected by adrenergic signaling which can reduce murine liver damage in a model of non-alcoholic steatohepatitis (35). In addition, better outcomes in patients with liver disease have been documented in patients with uninhibited adrenergic signaling (i.e. no β-blockers) (36). This may explain the specific decrease in liver GVHD in recipients in which adrenergic signaling is intact, while it appears that adrenergic signaling is not affecting GVHD in the intestines. Overall, these data reinforce the already understood importance of the effect of a physiological stress (i.e., as generated here through mild cold stress) on inflammatory diseases (7-9, 37).
Our data highlight the importance of ambient temperature as a variable that influences the baseline stress levels in mice and thus the outcome of experimental manipulation in mouse models. This could contribute to different outcomes between laboratories and an inability to duplicate experimental settings. Our studies have not addressed whether there could be differences in GVHD severity within the mandated range of ambient housing temperature (20-26°C) (38). In particular, if GVHD severity increases correspondingly within the range of 20-26°C it could help to explain some of the variation observed between labs or even within the same lab due to fluctuations in ambient temperature. This could also hinder the potential for translating preclinical findings to the clinic (39, 40). While patients may not typically experience chronic cold stress, the sympathetic response produces norepinephrine in response to many other types of stress which are experienced at widely varying degrees in patients (41). Thus, investigating multiple forms of stress may enhance our understanding of how therapies, especially immune based interventions (i.e. alloHCT), affect patients and the development of complications such as GVHD.
For future studies it will be important to understand the impact of β-AR signaling on graft versus tumor (GVT) effect. Tumor cells also express β-AR so direct effects of receptor agonists or norepinephrine on tumor progression are possible. In fact, we recently demonstrated that β-AR signaling induced by 22°C housing conditions or β-AR agonists activates anti-apoptotic signaling pathways (11). In addition, others have shown that β-AR antagonists prevent the anti-apoptotic effect of adrenergic stress (42, 43). This means that in addition to influencing GVHD severity, adrenergic stress can affect tumor resistance to cell death. Studies using different combinations of β-AR specific drugs, and use of β-AR knockout mice and adrenergic receptor deficient tumors lines should allow investigators to understand the complex role of β-AR signaling following transplantation. Overall, we expect this work to lead to studies employing β-agonists to prevent clinical GVHD. Equally as important, these data may lead to a more cautious use of non-specific β-AR antagonists (i.e., commonly prescribed β-AR blockers) for other unrelated medical indications during clinical alloHCT so as to avoid the potential risk for increased GVHD severity.
Acknowledgements
We thank Drs. Sarah Holstein, Bonnie Hylander, Michelle Messmer, Kelvin Lee and Michael Nemeth for their discussions and technical support. We thank Jeanne Prendergast for her laboratory and editorial assistance.
Footnotes
This research was supported by NIH Grants R01CA135368 (E.A.R.), R01CA184728 (X.C.) and T32 CA085183 (N.D.L., K.M.K., J.W.-L.E.). This work utilized Shared Resources supported by the Roswell Park Cancer Institute’s Comprehensive Cancer Center Support Grant CA016056.
Competing Financial Interests
The authors report no conflicts of interest.
References
- 1.Hahn T, McCarthy PL, Jr., Zhang MJ, Wang D, Arora M, Frangoul H, Gale RP, Hale GA, Horan J, Isola L, Maziarz RT, van Rood JJ, Gupta V, Halter J, Reddy V, Tiberghien P, Litzow M, Anasetti C, Pavletic S, Ringden O. Risk factors for acute graft-versus-host disease after human leukocyte antigen-identical sibling transplants for adults with leukemia. JClin Oncol. 2008;26:5728–5734. doi: 10.1200/JCO.2008.17.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Socie G, Blazar BR. Acute graft-versus-host disease: from the bench to the bedside. Blood. 2009;114:4327–4336. doi: 10.1182/blood-2009-06-204669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng D, Lewis D, Dejbakhsh-Jones S, Lan F, Garcia-Ojeda M, Sibley R, Strober S. Bone marrow NK1.1(−) and NK1.1(+) T cells reciprocally regulate acute graft versus host disease. The Journal of experimental medicine. 1999;189:1073–1081. doi: 10.1084/jem.189.7.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. The Journal of experimental medicine. 1978;148:1687–1698. doi: 10.1084/jem.148.6.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palathumpat V, Dejbakhsh-Jones S, Holm B, Strober S. Different subsets of T cells in the adult mouse bone marrow and spleen induce or suppress acute graft-versus-host disease. J Immunol. 1992;149:808–817. [PubMed] [Google Scholar]
- 6.Socie G, Blazar BR. Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation. Elsevier; USA: 2013. [Google Scholar]
- 7.Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, Sexton S, Hong CC, Gordon CJ, Abrams SI, Repasky EA. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20176–20181. doi: 10.1073/pnas.1304291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaskill B, Rohr S, Pajor E, Lucas J, Garner J. Some like it hot: Mouse temperature preferences in laboratory housing. Appl. Anim. Behav. Sci. 2009;116:279–285. [Google Scholar]
- 9.Gordon CJ. Thermal physiology of laboratory mice: Defining thermoneutrality. Journal of thermal biology. 2012;37:654–685. [Google Scholar]
- 10.Kokolus KM, Spangler HM, Povinelli BJ, Farren MR, Lee KP, Repasky EA. Stressful presentations: mild cold stress in laboratory mice influences phenotype of dendritic cells in naive and tumor-bearing mice. Frontiers in immunology. 2014;5:23. doi: 10.3389/fimmu.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eng JW-L, Reed CB, Kokolus KM, Pitoniak R, Utley A, Repasky EA, Hylander BL. Housing temperature-induced stress drives therapeutic resistance in murine tumor models through β2-adrenergic receptor activation. Nature communications. 2015 doi: 10.1038/ncomms7426. B. MJ, M. W.W. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herve J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, Blancou P. beta2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol. 2013;190:3163–3171. doi: 10.4049/jimmunol.1201391. [DOI] [PubMed] [Google Scholar]
- 13.Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5300–5305. doi: 10.1073/pnas.0808851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kin NW, Sanders VM. It takes nerve to tell T and B cells what to do. Journal of leukocyte biology. 2006;79:1093–1104. doi: 10.1189/jlb.1105625. [DOI] [PubMed] [Google Scholar]
- 15.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. The Journal of experimental medicine. 2014;211:2583–2598. doi: 10.1084/jem.20141132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16574–16579. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe A, Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J Immunol. 2000;165:680–690. doi: 10.4049/jimmunol.165.2.680. [DOI] [PubMed] [Google Scholar]
- 18.Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE. alpha1-adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. The Journal of pharmacology and experimental therapeutics. 2011;338:648–657. doi: 10.1124/jpet.110.178012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cellular signalling. 2007;19:251–260. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Ramer-Quinn DS, Baker RA, Sanders VM. Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production. J Immunol. 1997;159:4857–4867. [PubMed] [Google Scholar]
- 21.Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, Reis VO, Keller AC, Brum PC, Basso AS. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. European journal of immunology. 2013;43:1001–1012. doi: 10.1002/eji.201243005. [DOI] [PubMed] [Google Scholar]
- 22.Shlomchik WD. Graft-versus-host disease. Nature reviews. Immunology. 2007;7:340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 23.Ding X, Bian G, Leigh ND, Qiu J, McCarthy PL, Liu H, Aygun-Sunar S, Burdelya LG, Gudkov AV, Cao X. A TLR5 agonist enhances CD8(+) T cell-mediated graft-versus-tumor effect without exacerbating graft-versus-host disease. J Immunol. 2012;189:4719–4727. doi: 10.4049/jimmunol.1201206. [DOI] [PubMed] [Google Scholar]
- 24.Bian G, Ding X, Leigh ND, Tang Y, Capitano ML, Qiu J, McCarthy PL, Liu H, Cao X. Granzyme B-mediated damage of CD8+ T cells impairs graft-versus-tumor effect. J Immunol. 2013;190:1341–1350. doi: 10.4049/jimmunol.1201554. [DOI] [PubMed] [Google Scholar]
- 25.Heymer B. Clinical and Diagnostic Pathology of Graft-versus-Host Disease. Springer; 2002. [Google Scholar]
- 26.Chen JC, Chang ML, Muench MO. A kinetic study of the murine mixed lymphocyte reaction by 5,6-carboxyfluorescein diacetate succinimidyl ester labeling. Journal of immunological methods. 2003;279:123–133. doi: 10.1016/s0022-1759(03)00236-9. [DOI] [PubMed] [Google Scholar]
- 27.Merad M, Hoffmann P, Ranheim E, Slaymaker S, Manz MG, Lira SA, Charo I, Cook DN, Weissman IL, Strober S, Engleman EG. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nature medicine. 2004;10:510–517. doi: 10.1038/nm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka M, Yoshida M, Emoto H, Ishii H. Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: basic studies. European journal of pharmacology. 2000;405:397–406. doi: 10.1016/s0014-2999(00)00569-0. [DOI] [PubMed] [Google Scholar]
- 29.Baek K, Hwang HR, Park HJ, Kwon A, Qadir AS, Baek JH. Propranolol, a beta-adrenergic antagonist, attenuates the decrease in trabecular bone mass in high calorie diet fed growing mice. BMB reports. 2014;47:506–511. doi: 10.5483/BMBRep.2014.47.9.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, Baker EM, Cao YA, Contag CH, Negrin RS. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 2005;106:1113–1122. doi: 10.1182/blood-2005-02-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–415. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 32.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nature reviews. Immunology. 2012;12:443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsieh AC, Carlson LD. Role of the thyroid in metabolic response to low temperature. The American journal of physiology. 1957;188:40–44. doi: 10.1152/ajplegacy.1956.188.1.40. [DOI] [PubMed] [Google Scholar]
- 34.Depocas F. The calorigenic response of cold-acclimated white rats to infused noradrenaline. Canadian journal of biochemistry and physiology. 1960;38:107–114. [PubMed] [Google Scholar]
- 35.McKee C, Soeda J, Asilmaz E, Sigalla B, Morgan M, Sinelli N, Roskams T, Oben JA. Propranolol, a beta-adrenoceptor antagonist, worsens liver injury in a model of non-alcoholic steatohepatitis. Biochemical and biophysical research communications. 2013;437:597–602. doi: 10.1016/j.bbrc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serste T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, Moreau R, Lebrec D. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017–1022. doi: 10.1002/hep.23775. [DOI] [PubMed] [Google Scholar]
- 37.Maloney SK, Fuller A, Mitchell D, Gordon C, Overton JM. Translating animal model research: does it matter that our rodents are cold? Physiology (Bethesda) 2014;29:413–420. doi: 10.1152/physiol.00029.2014. [DOI] [PubMed] [Google Scholar]
- 38.Guide for the Care and Use of Laboratory Animals. 8th National Reserach Council; Washington, DC: 2011. [Google Scholar]
- 39.Francia G, Kerbel RS. Raising the bar for cancer therapy models. Nature biotechnology. 2010;28:561–562. doi: 10.1038/nbt0610-561. [DOI] [PubMed] [Google Scholar]
- 40.Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. American journal of translational research. 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- 41.Desborough JP. The stress response to trauma and surgery. British journal of anaesthesia. 2000;85:109–117. doi: 10.1093/bja/85.1.109. [DOI] [PubMed] [Google Scholar]
- 42.Zhang D, Ma Q, Wang Z, Zhang M, Guo K, Wang F, Wu E. beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Molecular Cancer. 2011:10. doi: 10.1186/1476-4598-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shan T, Ma Q, Zhang D, Guo K, Liu H, Wang F, Wu E. beta2-adrenoceptor blocker synergizes with gemcitabine to inhibit the proliferation of pancreatic cancer cells via apoptosis induction. European journal of pharmacology. 2011;665:1–7. doi: 10.1016/j.ejphar.2011.04.055. [DOI] [PubMed] [Google Scholar]