

Abstract
Interleukin-25 or IL-17E is a member of IL-17 cytokine family and has immune-modulating activities. The role of IL-25 in maintaining lipid metabolic homeostasis remains unknown. We investigated the effects of exogenous IL-25 or deficiency of IL-25 on hepatic lipid accumulation. IL-25 expression was examined in paraffin-embedded tissue sections of liver from patients or in the livers from mice. Mouse model of steatosis was induced by feeding a high-fat diet (HFD). Extent of steatosis as well as expression of cytokines, key enzymes for lipid metabolic pathways, markers for Kupffer cells/macrophages, and lipid droplet proteins were analyzed. Our results show that hepatic steatosis in mice was accompanied by increased lipid droplet proteins, but decreased IL-25 in the liver. Decreased hepatic IL-25 was also observed in patients with fatty liver. Administration of IL-25 to HFD-fed wild-type mice led to a significant improvement in hepatic steatosis. This effect was associated with increased expression of IL-13, development of alternatively-activated Kupffer cells/macrophages, and decreased expression of lipid droplet proteins in the liver. In contrast, administration of IL-25 to HFD-fed mice deficient in STAT6 or IL-13 had no effects. In addition, stimulation of primary hepatocytes with IL-13, but not IL-25, resulted in down-regulation of lipid droplet proteins. Finally, mice deficient in IL-25 had exacerbated hepatic lipid accumulation when fed the HFD. These data demonstrate that dysregulated IL-25 expression contributes to lipid accumulation, while exogenous IL-25 protects against hepatic steatosis through IL-13 activation of STAT6. IL-25 and IL-13 are potential therapeutic agents for hepatic steatosis and associated pathologies.
Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a spectrum of liver diseases ranging from simple steatosis, steatohepatitis, and cirrhosis without excessive consumption of alcohol (1). Major etiological factors that contribute to the development of NAFLD include obesity, type 2 diabetes, and dyslipidemia. As the incidence of obesity reaches endemic levels worldwide, the prevalence of NAFLD has been increasing steadily in recent decades with a current estimate of 20% of all adults being affected (1). The hallmark of NAFLD is steatosis characterized by a histologic manifestation of intracytoplasmic lipid accumulation in the form of triglycerides (TG) in hepatocytes. Excessive lipid accumulation/storage in the liver arises from an imbalance between lipid acquisition and disposal; therefore, dysregulated lipid metabolic pathways are the important determinants of hepatic steatosis (1). Inflammation is a core component linking obesity and associated metabolic diseases including NAFLD (2), and pro-inflammatory cytokines such as IL-1β, TNFα, and IL-6 are believed to be key mediators for metabolic dysregulation.
IL-25, also called IL-17E, is a member of the IL-17 cytokine family (3). While other members of the family, especially IL-17A and IL-17F, have biological activities similar to Th1 inflammatory cytokines, IL-25 promotes type 2 immunity. The receptor for IL-25 (IL-25R) is a heterodimer consisting of IL-17RB and IL-17RA (4). The downstream signaling pathway of IL-25 remains to be fully elucidated, although transcription factors Act1 and TRAF6 may be involved and the signaling events initiated by binding to IL-17RB are likely to be tissue/cell-specific (5). Recent studies demonstrated that IL-25 acts primarily on type 2 innate lymphoid cells (ILC2) to induce the production of the type 2 cytokines IL-13 and IL-5 (6). It is critical for host protective immunity against parasitic nematode infection and is associated also with airway allergic inflammation (7–9). Of equal importance is the ability of IL-25 to inhibit pro-inflammatory Th1 and Th17 cytokine responses that are implicated in the metabolic manifestation associated with obesity (10). Therefore, IL-25 is emerging as a key regulator of inflammation. The highest levels of IL-25 are found in the gastrointestinal tract and lung, where it is expressed primarily by epithelial cells and certain types of immune cells (8, 9, 11–13). More recently, a study showed that hepatocytes were capable of producing IL-25 and decreased hepatic IL-25 was observed in animals or humans with hepatitis (14). Whether IL-25 is involved in regulation of lipid metabolism and associated hepatic steatosis is not known.
Given that inflammation affects the progression of NAFLD whereas IL-25 possesses anti-inflammatory properties, we hypothesized that constitutively expressed IL-25 maintains lipid metabolic homeostasis while enhanced IL-25 production in vivo would protect against the excessive lipid accumulation in the liver. The present study was designed to investigate: (i) the effects of exogenous IL-25 on obesity-associated hepatic steatosis in mice; (ii) the molecular mechanisms of IL-25 regulation of lipid accumulation/storage; (iii) the contribution of the IL-13-STAT6 axis to the effects of IL-25; and (iv) the impact of IL-25 deficiency on expression of hepatic steatosis.
Materials and Methods
Mice and diet
C57BL/6 wild type (WT) mice were purchased from National Cancer Institute Mouse Repository (Frederick, MD). Mice deficient in STAT6 (STAT6−/−, Jackson laboratory) or IL-13 (IL-13−/−, National Institutes of Allergy and Infectious Diseases Taconic contract) on C57BL/6 background were bred in the USDA/Beltsville animal facility. Mice deficient in IL-25 (IL-25−/−) were generated by Regeneron Pharmaceuticals (Tarrytown, NY) and were backcrossed to C57BL/6 mice for 10 generations. A commonly used high-fat diet (HFD) fed mouse model of hepatic steatosis was selected because it is considered physiologically relevant. Male, 5-week-old mice were fed a HFD (60% of kcal from fat, Research Diet, New Brunswick, NJ) or normal control diet (NCD, 10% of kcal from fat) ad libitum. These studies were conducted with institutional approval from both the University of Maryland, Baltimore and the USDA Beltsville Area Animal Care and Use Committees (protocol #13-003), in accordance with principles set forth in the Guide for Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, National Research Council, Health and Human Services Publication.
Administration of IL-25
After being on the respective diet for ~14 weeks, mice were separated into treatment or control groups. Mice in the treatment group were injected i.p. with 1μg of mouse recombinant IL-25 (R&D Systems, Minneapolis, MN) in 100μl of PBS daily for five days in the first week followed by three injections per week for an additional two weeks. Control mice were given injections of 35μg BSA, which is equal to the amount of BSA carrier protein included in the IL-25 preparation. The amount of cytokine administered was based on the minimum dose of IL-25 that induced a prominent Th2 immune response (8).
Mouse hepatocyte preparation and treatment
Mouse primary hepatocytes were isolated from HFD-fed obese mice using a two-step collagenase perfusion method (15). Hepatocytes were seeded at 1 × 106 cells/well in 6-well collagen coated plate in DMEM supplemented with 5% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 4 μg/ml insulin, and 1 μM dexamethasone. After 4 hours of attachment at 37°C in a humidified atmosphere of 5% CO2, the medium was changed to DMEM without FBS and cells were cultured for another 24 hours before treatment with IL-25 or IL-13. Medium was replaced on a daily basis.
RNA extraction, cDNA synthesis, real-time quantitative PCR (qPCR), and ELISA
Total RNA was extracted with TRIzol reagent (Invitrogen, Grand Island, NY) per the manufacturer’s instructions. RNA samples (2μg) were reverse-transcribed to cDNA using the First Strand cDNA Synthase Kit (MBI Fermentas, Hanover, MD) with random hexamer primer. qPCR was performed on an iCycler detection system (Bio-Rad, CA) in a 25μl volume using SYBR green Supermix (Bio-Rad, Hercules, CA). Amplification conditions were: 95°C for 3 min, 50 cycles of 95°C for 15s, 60°C for 15s, and 72°C for 20s. The fold change in mRNA expression for targeted gene was relative to the respective vehicle after normalization to 18s rRNA. Tissue homogenates of liver samples were prepared with RIPA buffer (Cell signaling technology, Beverly, MA). IL-25 protein expression in the liver was measured with ELISA kit according to the manufacturer’s instructions (BioLegend, San Diego, CA).
Hematoxylin-eosin (H&E) and immunofluorescent stainingon mouse liver sections
Tissue sections (5μm) were cut from frozen blocks of mouse liver and stained with H&E for histological analysis. Slides were graded for steatosis according to the Kleiner system by an investigator who was unaware of the treatment and mouse genotype (16). A score of 0–3 was assigned to describe the extent of steatosis based on lipid accumulation in the hepatocytes (0, <5%; 1, 5–33%; 2, 33–66%; 3, >66%). Immunofluorescent staining of mouse liver sections was carried out as described previously (17). Briefly, the slides were blocked with 5% normal donkey serum and then incubated with anti-F4/80 (BioLegend, San Diego, CA) and anti-YM-1 (R&D Systems, Minneapolis, MN) antibodies overnight. The slides were stained with Dylight 488–donkey anti-rat IgG and Dylight 659–donkey anti-goat IgG (1:400; Jackson ImmunoResearch, West Grove, PA) for 1 hour and then digitally photographed with a Nikon TE 2000-E microscope (Melville, NY). The images were taken by establishing settings for the samples from mice injected with BSA and using the same conditions to evaluate the samples from mice injected with IL-25. Comparisons were made only among slides prepared on the same day.
Immunohistochemical staining on human liver sections
Paraffin-embedded tissue slides of liver sections were acquired from a total of 14 patients. Six of them had hepatic steatosis, diagnosed initially with ultrasound examination and later confirmed by liver biopsy evaluation. The other eight patients had no hepatic steatosis and were used as controls. The medical history was reviewed retrospectively by a medical doctor specialized in the gastroenterology and hepatology. All of the patients were >18 year old at the time of biopsy, and had no history of alcohol abuse, serologic evidence of viral hepatitis, blood transfusion, or history of other competing etiologies for hepatic steatosis and co-existing causes for chronic liver disease (18). For immunohistochemical staining, slides were dewaxed with xylene, gradiently washed with ethanol, blocked with goat serum, and then incubated with anti-IL25 mono-antibody (1:100, R&D Systems, Minneapolis, MN, Cat. No. MAB1258) or the isotype control. The slides were stained with Peroxidase/DAB kit (Dako Denmark A/S, Denmark) and then digitally photographed with OLYMPUS BX51WI microscope.
Lipid extraction and analysis
Tissue homogenates were prepared in lipid extraction buffer, shaken in the dark for 2 hours, and centrifuged at 3,500 rpm for 10 min. The soluble portion was transferred to a 1.5-ml tube and dried under vacuum for 30 min. The dried lipids were made soluble in TG assay buffer by vortexing until the lipids were homogeneous. Levels of hepatic TG were determined using a commercial kit (Cayman Chemical, Ann Arbor, MI).
Western-blot analysis
Western-blot analysis was performed as described previously (19). In brief, tissue lysates prepared in RIPA buffer (Cell Signaling Technology, Beverly, MA) were separated on 14% Novex Tris-glycine Mini Gels and transferred to Novex Nitrocellulose Membrane (Life Technologies, Grand Island, NY). The membranes were blocked with 5% milk and incubated with primary antibody against cell death-inducing DFFA-like effector A (CIDEA, 1:2000, Cell Signaling Technology, Beverly, MA) overnight. Bands were detected using a HRP-conjugated goat anti-rabbit secondary antibody (Santa Cruz, CA, USA). The Western-blot was re-probed with anti-GAPDH as a loading control.
Data analysis
Statistical analysis was performed using one-way ANOVA followed by Neuman-Keuls test to compare the difference among three or more treatment groups, or the Student t test to compare the difference between two groups. P values of <0.05 were considered significant.
Results
HFD-induced hepatic steatosis is associated with decreased expression of IL-25 in the liver
Hepatocytes are capable of producing IL-25 and decreased hepatic IL-25 expression has been reported in humans or mice with hepatitis (14). We established a HFD-induced model of hepatic steatosis in mice and evaluated a role of IL-25 in NAFLD. As expected, C57BL/6 mice fed the HFD for 14 weeks had significantly enlarged livers and increased levels of hepatic TG (Fig. 1A & B), characteristics of steatosis, when compared with age- and sex-matched mice fed NCD. Evaluation of the H&E-stained liver sections indicated that HFD-fed mice had high degree of steatosis accompanied by the presence of both large and small fat vacuoles (Fig. 1C). In addition, steatotic livers from HFD-fed mice had significantly up-regulated gene expression of lipid droplet (LD)-associated proteins including the CIDE proteins Cidea, Cideb, Cidec as well as perilipin 2 (Plin2) (Fig. 1D). Hepatic expression of F4/80 (Emr1) and Cd11b (Fig. 1E), the markers for Kupffer cell (20, 21), were comparable between mice fed the NCD and HFD. A significant up-regulation of Tnfα and Ccl2, but not Il6, was also observed in the livers of HFD-fed mice (Fig. 1E). Interestingly, HFD-fed mice had a significantly decreased IL-25 protein in the livers when compared to NCD-fed mice (Fig. 1F).
Figure 1. Increased expression of Lipid droplet (LD)-associated proteins and decreased expression of IL-25 in the livers of mice with steatosis.
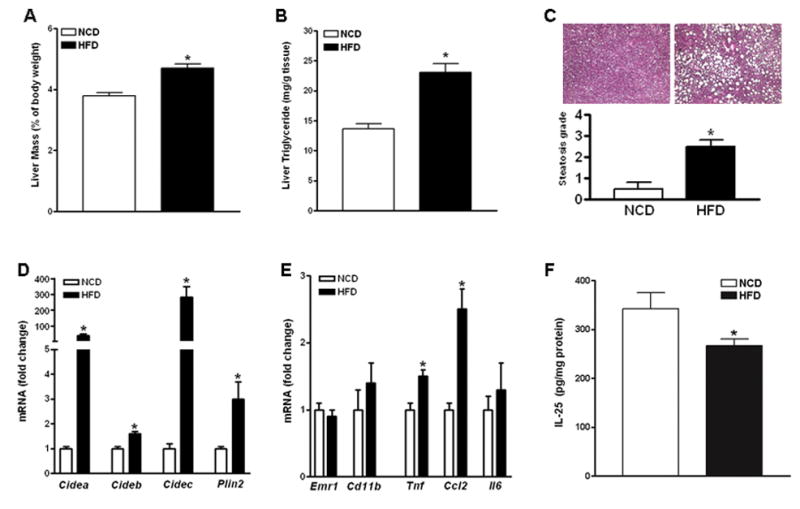
Mice were fed a high-fat diet (HFD) or normal control diet (NCD). (A) Liver mass; (B) Liver triglyceride levels; (C) H&E-stained liver sections (100X) and steatosis scores; (D) Hepatic expressions of LD-associated proteins by qPCR; (E) Hepatic expressions of macrophage markers and immune mediators by qPCR. (F) Levels of IL-25 protein expression in the liver by ELISA. Data shown in bar graphs are mean ± SEM and are representatives of two independent experiments with five-ten mice per group per experiment. * P<0.05 versus NCD.
Exogenous IL-25 ameliorates high-fat diet-induced hepatic steatosis
We next determined if exogenous IL-25 ameliorates HFD-induced hepatic steatosis. To this end, mice were fed the HFD for ~14 weeks and then administered with either IL-25 or BSA. After a three-week treatment, mice receiving IL-25 had significantly reduced hepatic steatosis indicated by decreased liver mass and hepatic TG levels (Fig. 2A & B). The amelioration was further confirmed by histological evaluation of H&E-stained liver sections showing decreased size/number of fat vacuoles and lower steatosis score (Fig. 2C & D). In addition, IL-25 treatment did not alter significantly the liver mass or TG levels in mice fed the NCD (Fig. 2). Of note, IL-25 decreased food intake in both groups of NCD- and HFD-fed mice in the first week of injection, but induced loss of body weight and decrease in hepatic lipid accumulation only in HFD-fed mice (Fig. S1). This suggests that the transient decrease in food intake was unlikely a major factor for the loss of body weight or the amelioration of hepatic steatosis in IL-25-treated HFD-fed mice.
Figure 2. Exogenous IL-25 ameliorates HFD-induced hepatic steatosis.
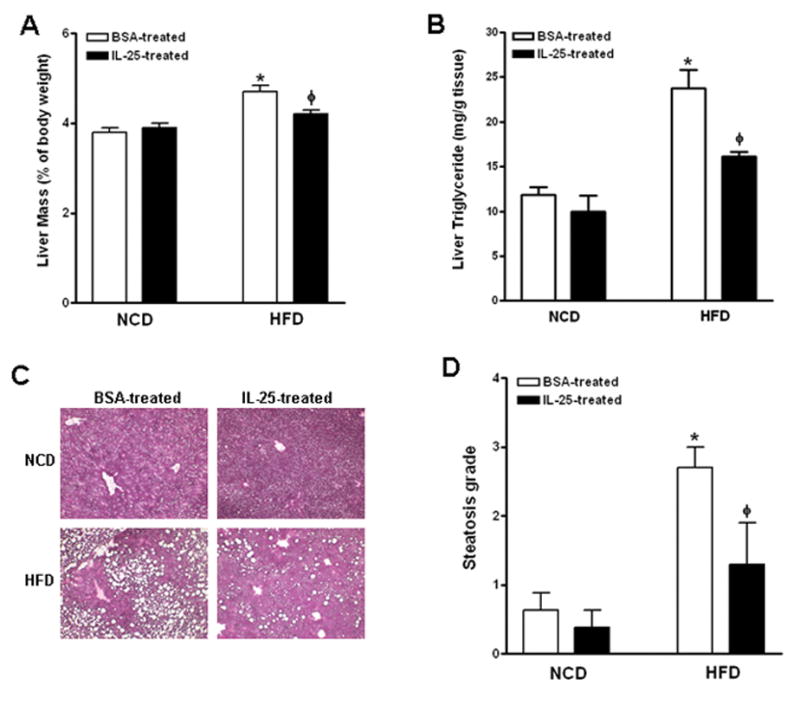
Mice were fed the high-fat diet (HFD) or normal control diet (NCD) and then injected with IL-25 or BSA. (A) Liver mass; (B) Liver triglyceride levels; (C) H&E-stained liver sections (x100); (D) Steatosis scores. Data shown in bar graphs are mean ± SEM and are representatives of two independent experiments with five mice per group per experiment. * P<0.05 versus respective NCD; ϕ P<0.05 versus respective BSA-treated.
To dissect the lipid metabolic pathways that might contribute to IL-25-induced improvement in hepatic steatosis, we analyzed expression of genes encoding key enzymes for lipid metabolism. IL-25 treatment did not alter hepatic expression of key lipogenic enzymes (Fasn, Acly, and Acaca), lipid transporters (Cd36, Fatp2, and Fatp5), hepatic lipase (Lipc), or carnitine palmitoyltransferase 1a (Cpt1a), whereas it significantly increased the expression of hydroxyacyl-Coenzyme A dehydrogenase (Hadh), an enzyme important for fatty acid (FA) oxidation (Fig. 3A & B, and data not shown). In addition, IL-25 treatment significantly decreased expressions of the CIDEs as well as Plin2 in the livers of HFD-fed mice (Fig. 3C). The decrease in CIDEA protein in the livers of IL-25-treated mice was further confirmed by Western-blot (Fig. 3D). Hepatic expression of these LD-associated proteins was not significantly altered by IL-25 treatment in NCD-fed mice, possibly due to the low constitutive expression of these genes (data not shown). These data suggest that increased FA oxidation and altered LD formation may contribute to the effects of IL-25 on hepatic steatosis.
Figure 3. Administration of IL-25 to HFD-fed mice alters the expressions of key enzyme for lipid metabolic pathway and LD-associated proteins in the liver.
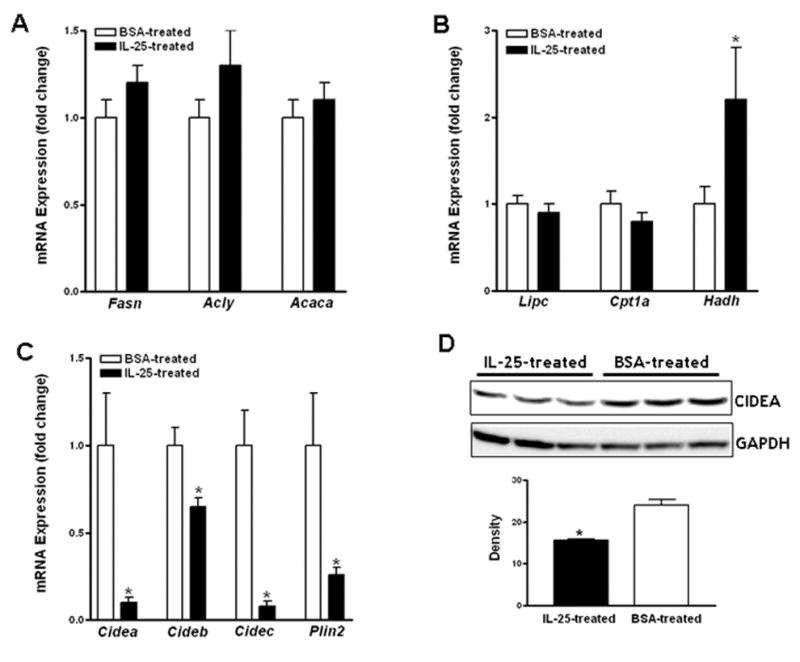
Mice were fed the high-fat diet (HFD) or normal control diet (NCD) and then injected with IL-25 or BSA. qPCR was carried out to examine gene expression of lipogenic enzymes (A), fatty acid oxidation proteins (B), and lipid droplet-associated proteins (C). CIDEA protein was detected by Western-blot and the density represents average of the bands after normalization to GAPDH (D). Data shown in bar graphs are mean ± SEM and are representatives of two independent experiments with five mice per group per experiment. * P<0.05 versus respective BSA-treated.
Exogenous IL-25 induces expression of type 2 cytokine and alternative activation of Kupffer cell/macrophage in the liver
Consistent with a role of IL-25 in promoting type 2 immunity, there was an up-regulation of major type 2 cytokine gene expression of Il13 and Il5 in the livers of HFD-fed mice receiving IL-25 (Fig. 4A). Type 2 innate lymphoid cells (ILC2) are the major IL-25-responsive/IL-13-producing cells in the liver (22). Accordingly, IL-25 injection increased the expression of major ILC2 markers (6) including CD25 (Il2ra), CD90 (Thy1), CD127 (Il7r), and Sca-1 (Ly6a) as well as the transcription factor RORα in the liver (Fig. 4A). Other type-2 related cytokines Il4, Il25, and Il33 were expressed either at relatively low levels in the liver or unaffected by treatment with IL-25 (data not shown). In contrast, IL-25 treatment significantly decreased the expression of Ccl2, but not Tnfα or Il6, in the livers of HFD-fed mice (Fig. 4B). Up-regulation of Il5 and Il13 as well as a down-regulation of Ccl2 were also detected in the livers of IL-25-treated NCD-fed mice (Fig. S2A).
Figure 4. Administration of IL-25 to HFD-fed mice induces a type 2 cytokine response and alternative activation of Kupffer cell/macrophage in the liver.
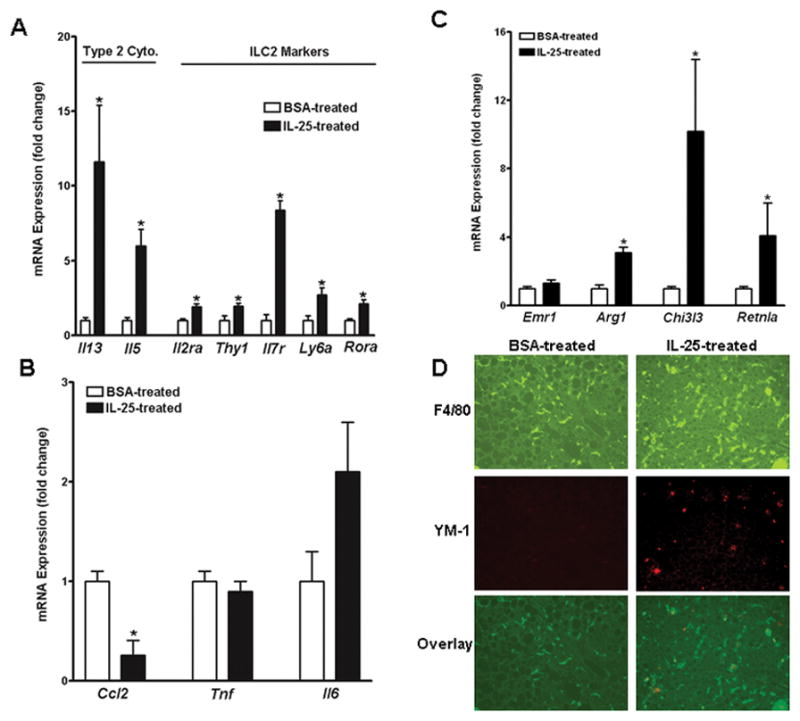
Mice were fed a high-fat diet (HFD) and then received injections of IL-25 or BSA. (A) Hepatic expression of type 2 cytokines and molecular markers for ILC2; (B) Hepatic expression of markers for Kupffer cell/macrophage activation. (C) Immunofluorescent staining of liver sections with anti-F4/80 (green) and anti-YM-1 (red). The images are representatives of five mice in each group. Data shown in bar graphs are mean ± SEM and are representatives of two independent experiments with five mice per group per experiment. *P<0.05 versus respective BSA-treated.
Kupffer cells in the liver are derived from either intrahepatic precursors or infiltrating monocytes (23), and play an important role in obesity-induced hepatic steatosis (24). IL-25 treatment did not affect the gene expression of F4/80 (Emr1), but significantly up-regulated the expression of alternatively-activated (M2) Kupffer cell markers arginase I (Arg1), YM-1 (Chi3l3), and FIZZ1 (Retnla) (Fig. 4C). Increased number of M2 Kupffer cells/macrophages in the liver was validated further by immunofluorescent staining with anti-YM-1, as more F4/80+/YM-1+ cells could be visualized in the livers from IL-25-treated HFD-fed mice than in those from BSA-treated mice (Fig. 4D). Likewise, M2 markers were up-regulated by IL-25 administration in the livers of NCD-fed mice (Fig. S2B).
Amelioration of hepatic steatosis induced by IL-25 depends upon STAT6 and IL-13
The increased expression of IL-13 and number of alternatively-activated (M2) Kupffer cells/macrophages in the livers of HFD-fed mice injected with IL-25 prompted us to investigate whether IL-13-STAT6 axis mediated the beneficial effects of IL-25. Thus, WT and STAT6−/− mice were fed the HFD or NCD for 14 weeks and then treated with IL-25 or BSA. IL-25 administration to either NCD- or HFD-fed STAT6−/− mice had no effect on liver mass or hepatic TG levels (Fig. 5A & B), while HFD-fed WT mice treated with IL-25 showed a marked reduction in liver mass and hepatic TG levels (data not shown and Fig. 2). IL-25 injection also did not impact histological steatosis grade of HFD-fed STAT6−/− mice (Fig. 5C). qPCR showed that IL-25 injection induced up-regulation of Il5 and IL13 in the livers of HFD-fed STAT6−/− mice (Fig. 5D) comparable to that of WT mice (Fig. 4A). However, the IL-25-induced up-regulation of M2 Kupffer cell markers, arginase I (Arg1) and YM-1 (Chi3l3), in WT mice (Fig. 4B & C) was almost absent in STAT6−/− mice (Fig. 5D) consistent with a STAT6-dependence. Immunofluorescent staining with anti-YM-1 confirmed no increase in the number of M2 Kupffer cells in the livers of IL-25-treated-STAT6−/− mice (data not shown). In addition, the IL-25-induced alterations in hepatic expression of Cideb, Cidec, Plin2, Hadh, or Ccl2 in WT mice (Fig. 3C & 4A) were absent in STAT6−/− mice, with the exception of Cidea which remained down-regulated (Fig. 5E and S3).
Figure 5. STAT6 dependence of IL-25-induced changes in hepatic steatosis and gene expression in the liver.
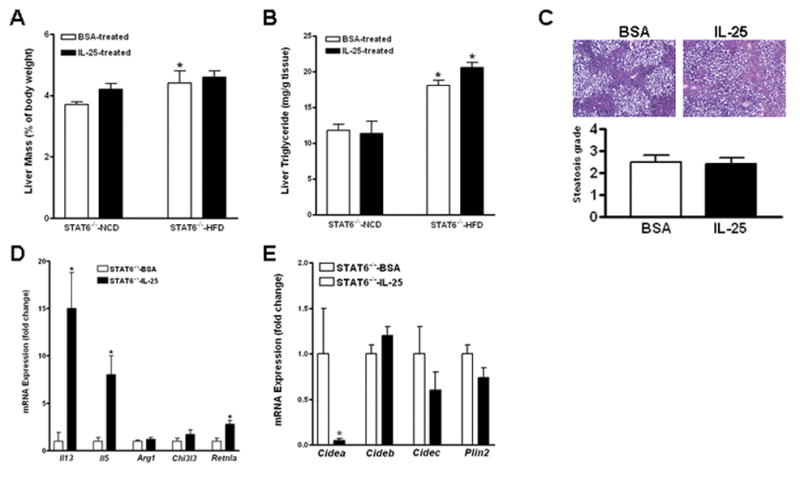
STAT6−/− mice were fed a high-fat (HFD) or normal control (NCD) diet and then treated with IL-25 or BSA. (A) Liver mass; (B) Liver triglyceride levels; (C) H&E-stained liver sections from HFD-fed mice (100X) and the steatosis scores; (D) Expression of cytokines or M2 Kupffer cell markers in the livers of HFD-fed mice; (E) Expression of LD-associated proteins in the livers of HFD-fed mice. Data shown in bar graphs are mean ± SEM and are representatives of two independent experiments with five mice per group per experiment. *P<0.05 versus respective STAT6−/−-NCD (A, B) or STAT6−/−-BSA (D, E).
Exogenous IL-25 can increase the expression of IL-4 and IL-13 (9, 10), both of which activate the STAT6 pathway, therefore, parallel experiments were carried out to determine the contribution of IL-13 to the effects of IL-25 on hepatic steatosis. In IL-13−/− mice fed the HFD for 14 weeks, IL-25 treatment for three weeks had no effect on liver mass, hepatic triglyceride level, or lipid accumulation (Fig. 6A & B). There was also no increase in the number of M2 Kupffer cells in the livers of IL-25-treated IL-13−/− mice, indicated by the similar transcript levels of the markers (Arg1, Retnla) between mice injected with BSA and IL-25 (Fig. 6C) and confirmed by immunofluorescent staining of liver section with anti-YM-1 (data not shown). Among the LD-associated proteins in the livers of IL-13−/− mice, IL-25 did not affect the expression of Cideb and Plin2, but surprisingly induced a modest up-regulation of the expression of Cidea and Cidec (Fig. 6D) contrary to that observed in WT mice (Fig. 3C).
Figure 6. Critical role of IL-13 in IL-25-induced changes in hepatic steatosis and gene expression in the liver.
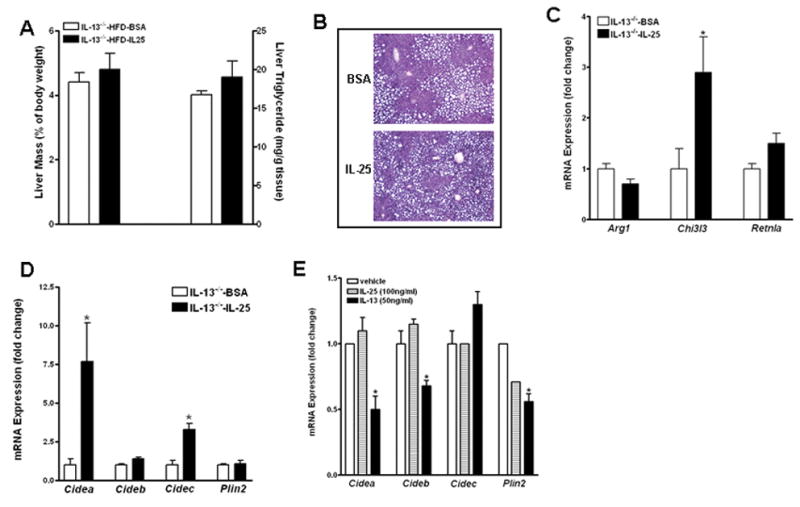
IL-13−/− mice were fed the high-fat diet (HFD) and then treated with IL-25 or BSA. (A) Liver mass and liver triglyceride levels; (B) H&E-stained liver sections from HFD-fed mice (x100); (C) Expression of M2 Kupffer cell markers in the livers of HFD-fed mice; (D) Expression of LD-associated proteins in the livers of HFD-fed mice; (E) Expression of LD-associated proteins in cultures of mouse primary hepatocytes. Data are mean ± SEM and are from one experiment with five mice per group (A-D) or representative of two independent experiments in triplicates (E). * P<0.05 versus IL-13−/−-BSA or vehicle.
The in vivo results indicated that IL-13-STAT6 axis plays a critical role in the IL-25-induced amelioration of hepatic steatosis likely through inhibiting expression of LD-associated proteins. Whether IL-13 directly acts on hepatocytes was examined further in primary culture of mouse hepatocytes. qPCR analysis showed that IL-13 treatment of hepatocytes significantly down-regulated the expression of Cidea, Cideb, and Plin2, while had no effect on the expression of Cidec (Fig. 6E). Of note, IL-13 treatment did not alter the gene expression of the LD-associated proteins described above in the hepatocytes isolated from STAT6−/− mice (data not shown). In contrast, IL-25 had no direct effect on the expression of any of the LD-associated proteins examined (Fig. 6E).
Genetic deletion of IL-25 increases susceptibility to HFD-induced steatosis
The potent effects of exogenous IL-25 on HFD-induced hepatic steatosis led us to examine further whether endogenous IL-25 is important for maintaining lipid metabolic homeostasis. Thus, WT and IL-25−/− mice were fed the HFD or NCD for ~16 weeks. When compared to age- and sex-matched WT mice, both NCD- and HFD-fed IL-25−/− mice had lower body weights (Fig. 7A). No significant differences in the liver mass or hepatic TG levels were detected between the two strains of mice fed the NCD (Fig. 7B & C). However, HFD-fed IL-25−/− mice had enlarged livers that were macroscopically pale in color (Fig. 7B & D) and increased levels of hepatic TG (Fig. 7C) as compared to their WT counterparts, although histological analysis was unable to reveal a significant difference statistically in steatosis (Fig. 7E) because of insufficient sensitivity of the grading system. The exacerbated hepatic steatosis in HFD-fed IL-25−/− mice was associated with up-regulated expressions of Cidea, Cideb, Cidec as well as Plin2 (Fig. 7F).
Figure 7. Mice with genetic deletion of IL-25 are more susceptible to HFD-induced hepatic steatosis.
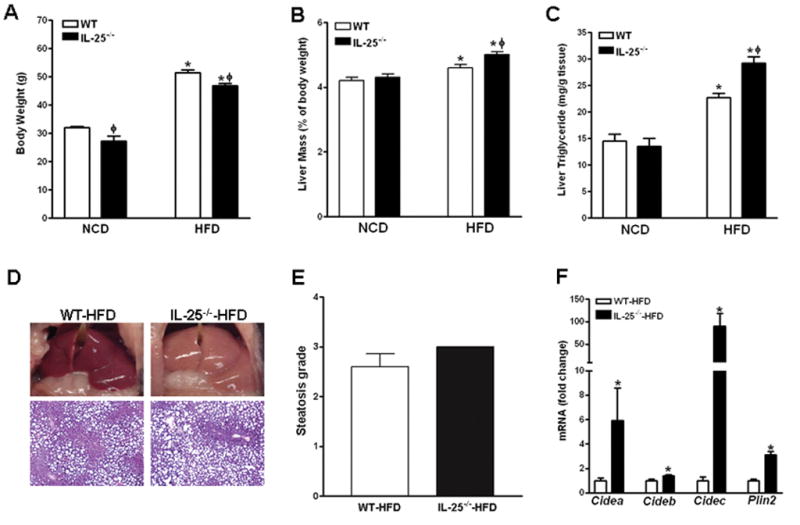
WT and IL-25−/− mice were fed a high-fat (HFD) or normal control (NCD) diet. (A) Body weight; (B) Liver mass; (C) liver triglyceride level; (D) Gross appearance of the liver, representative H&E-stained liver sections; (E) steatosis scores; (F) Hepatic expression of lipid droplet-associated proteins by qPCR. Data are mean ± SEM and are representatives of two independent experiments with five-eight mice per group. * P<0.05 versus respective NCD (A–C) or WT-HFD (F); ϕ P<0.05 versus respective WT.
Hepatic IL-25 expression is reduced in patients with fatty liver
To investigate the clinical relevance of our findings from mouse studies, we performed immunohistochemical staining with anti-IL-25 on paraffin-embedded liver sections from patients with or without fatty liver. IL-25-positive hepatocytes were visualized clearly in the liver sections from both groups of patients (Fig. 8). However, IL-25 staining was dramatically less in patients with fatty liver compared to those without. This is consistent with the ELISA results (Fig. 1F) showing decreased IL-25 protein in the livers of HFD-fed mice.
Figure 8. IL-25 expression is decreased in the livers of patients with hepatic steatosis.
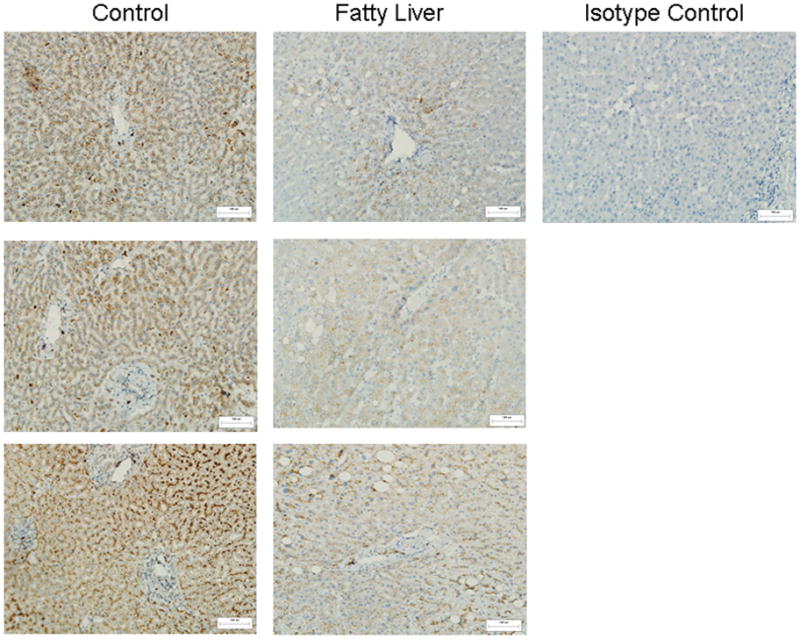
Immunohistochemical staining with anti-IL-25 antibody was performed on paraffin-embedded tissue slides of human liver sections. The images are representatives of six patients with hepatic steatosis and eight patients without hepatic steatosis as control (200x original magnification). One image from the staining with anti-IL-25 isotype control is also shown.
Discussion
Immune components play an important role in the development of hepatic steatosis, yet much of the focus has been on pro-inflammatory cytokines with less attention to anti-inflammatory immune mediators. Type 2–related cytokines, including IL-25, are capable of counter-regulating Th1 and Th17 immunity. Results from our current study indicate that exogenous IL-25 potently ameliorates the hepatic steatosis induced by HFD feeding through IL-13 activation of STAT6, whereas genetic deletion of IL-25 renders mice more susceptible to HFD-induced lipid accumulation in the liver. These results reveal a previous unrecognized role for IL-25 in the regulation of lipid metabolism.
IL-25 belongs to the IL-17 cytokine family (3). IL-17A, another key member of the family, was identified as a central player to the development and progression of NAFLD to steatohepatitis (25–27). Our results, however, show that IL-25 has beneficial effects against NAFLD, which is consistent with the established anti-inflammatory role for IL-25. Gut and lung epithelial cells are the recognized cellular sources for IL-25 (9, 10). Several types of immune cells may also produce IL-25 under certain circumstances, including Th2 cells, mast cells, and macrophages (8, 11–13). A recent study further identified hepatocytes as IL-25-expressing cells (14). Our current study showed that lipid accumulation in the liver was accompanied by decreased levels of IL-25 protein indicative of a role in regulation of lipid metabolism. Indeed, administration of IL-25 to HFD-fed WT mice resulted in a significant improvement in hepatic steatosis. Lipid accumulation in the liver arises from the imbalance of lipid acquisition and disposal, therefore, impaired FA oxidation, decreased lipid export, and increased de-novo lipogenesis in the liver are considered the key determinants of hepatic steatosis. Our results showed that IL-25 treatment did not affect hepatic expressions of key enzymes for lipogenesis or lipid export, but increased the expression of Hadh, an enzyme critical for FA oxidation, implicating one of the potential mechanisms for amelioration of steatosis by IL-25.
Lipid droplets are cytosolic lipid storage organelles present in nearly all cell types, and abnormal LD formation contributes to various human diseases including NAFLD (28). In hepatocytes, the size and number of TG-containing LDs are regulated by several types of proteins referred to as LD-associated proteins including members of CIDE and perilipin families. Both CIDEs and PLINs, especially the highly expressed PLIN2 in the liver, are believed to be the important regulators of lipid storage and formation of LDs in hepatocytes (29). Multiple lines of research demonstrated that mice or humans with hepatic steatosis have increased expression of LD-associated proteins in the liver (28). In addition, mice deficient in either one of the CIDEs exhibit decreased lipid accumulation and reduced hepatic steatosis when fed a HFD. In contrast, over-expression of CIDEA in mouse liver leads to increased hepatic lipid accumulation (30). Mice with PLIN2 deficiency are protected also from HFD-induced fatty liver disease accompanied by decreased adiposity (31). Consistent with previous observations, we observed that HFD-induced hepatic steatosis was associated with increased expression of all members of CIDE family and PLIN2. More importantly, treatment with IL-25 ameliorated hepatic steatosis and was accompanied by significantly decreased gene expression of Cidea, Cideb, Cidec, and Plin2. Conversely, IL-25−/− mice had increased hepatic levels of LD-associated proteins. These results strongly support that IL-25 regulates the expression of LD-associated protein, thereby controlling lipid accumulation in the liver.
Kupffer cells/macrophages in the liver represent ~10% of the resting total liver cell population and >80% of all tissue macrophages in the body (32). Traditional roles of Kupffer cells include launching biochemical attack, recruiting immune cells to the liver, eliminating cell debris and microorganisms, as well as antigen presentation. Kupffer cells are implicated in the pathogenesis of various liver diseases including hepatitis, alcoholic liver disease, and liver fibrosis (32). Like all macrophages, Kupffer cells undergo distinct pathways of activation and display different phenotypes depending on the cytokine microenvironment. Classically-activated (M1) Kupffer cells are induced by Th1 cytokines and are characterized by up-regulation of inducible nitric oxide synthase, whereas M2 Kupffer cells are induced by Th2 cytokines, IL-4 and IL-13, and feature highly up-regulated arginase I as well as the secretion of chitinase and “found in inflammatory zone” (FIZZ) family members such as YM1, FIZZ1, and FIZZ2 (33). Emerging evidence indicates that differentially-activated Kupffer cells play distinctive roles in the progression of NAFLD. Specifically, M1 Kupffer cells produce pro-inflammatory cytokines, such as IL-1β and TNFα, which inhibit the PPARα activity leading to decreased expression of genes involved in FA oxidation (24, 34). Depletion of Kupffer cells with clodronate liposomes reduces HFD-induced steatosis (24, 34). In contrast, alternatively-activated M2 Kupffer cells promote M1 Kupffer cell apoptosis through activation of arginase, thereby protecting against NAFLD (35). Our results showed that mice treated with IL-25 exhibited a polarized type 2 immunity characterized by up-regulations of IL-5 and IL-13 which led to development of M2 Kupffer cells in the liver. Thus, it is conceivable that the beneficial effects of IL-25 on steatosis are derived, at least in part, through its ability to promote a type 2 environment particularly increasing IL-13 that can lead to development of M2 Kupffer cells as demonstrated previously (36, 37). It should be noted that Kupffer cells/macrophages in the liver are a heterogeneous population (23). It remains to be determined whether the IL-25-induced M2 development occurred in the resident Kupffer cells, the infiltrated monocytes/macrophages, or both.
IL-25 induces increased expression of IL-4 and IL-13 that primarily activate the STAT6 pathway, as well as IL-5 that elicits eosinophilia (9, 38). Consistent with these observations, mice treated with IL-25 had increased gene expression of il5 and il13 but negligible levels of il4 in the liver. Notably, the amelioration of hepatic steatosis induced by exogenous IL-25 depended on STAT6 or IL-13, indicating that IL-25 acts through IL-13 activation of STAT6 signaling to regulate lipid accumulation in the liver. Hepatocytes are known to express receptors for IL-13 and IL-13 treatment of hepatocyte in culture can induce STAT6 activation (39–41). Our current studies showed that IL-13, but not IL-25, decreased the expression of LD-associated proteins in culture of hepatocytes from WT but not STAT6−/− mice, indicating that at least part of the effects of IL-25 on steatosis were mediated by its downstream IL-13 acting on hepatocytes through STAT6-dependent pathways. In addition, in vivo IL-25 treatment induced development of M2 Kupffer cells in the livers of HFD-fed WT but not in STAT6−/− and IL-13−/− mice. Coupled with the well-documented role of IL-13 in promoting M2 macrophages, it is plausible that IL-25 in the liver regulates the lipid accumulation/storage primarily through the direct action of IL-13 on Kupffer cells as well as on hepatocytes. What is somewhat surprising is that IL-25 administration to IL-13−/− mice induced a modest up-regulation of Cidea and Cidec. It is well recognized that IL-25 has multifaceted biological activities through the induction of different immune mediators. It is possible that some of the immune mediators up-regulate members of the CIDE family similarly to that by dietary FA shown previously (30), but the effects are counter-regulated when IL-13 signaling is intact. Despite the vital importance of IL-13 in the effects of IL-25, we were unable to detect significant differences in liver mass, hepatic triglyceride level, and steatosis grade among HFD-fed WT, STAT6−/−, and IL-13−/− mice. There were also no significant differences in mRNA expression for most of genes encoding LD-associated proteins among HFD-fed WT, STAT6−/−, and IL-13−/− mice (data not shown). These results suggest that the low constitutive level of IL-13 is not pivotal in controlling hepatic lipid accumulation. The major cellular source of IL-13 in the liver could be ILC2 that are known to respond to IL-25 (42). Several other types of immune cells are also capable of producing IL-13 including Th2 cells, eosinophils, NKT cells, and B cells (41), however, none of them are known to respond to IL-25 in vivo (42). Thus, it is likely that ILC2 are the IL-13-producing cells that mediate the IL-25 effects on hepatic steatosis.
Mice deficient in IL-25 develop intestinal inflammation in response to enteric nematode infection, characterized by increased expression of IL-17A as well as defective type 2 immune response (10). Hepatocytes can make IL-25 and decreased expression of IL-25 in the liver is observed in animals or human with hepatitis (14). Our results show that there is an increased severity of steatosis in mice with IL-25 deficiency accompanied by high levels of LD-associated proteins. Finally, the observation that patients with NAFLD had decreased immuno-staining of IL-25 in the livers demonstrates the clinical importance of hepatic expression of IL-25. The mechanisms by which IL-25 was decreased in steatosis remain to be investigated; however, the decrease in IL-25 was accompanied by an increase in TNFα in steatotic livers and TNFα was shown to negatively regulate IL-25 expression in the colon or brain (43). More studies are needed to ascertain whether this same mechanism of TNFα regulating IL-25 occurs in the liver.
Taken together, our study is the first to show that IL-25 is important in maintaining lipid metabolic homeostasis in the liver. Even though the detailed mechanism remains to be fully understood, it is believed that IL-25 in the liver stimulated ILC2 to release IL-13 that in turn acted on hepatocytes as well as Kupffer cells via STAT6-depedent pathways, which then altered the expression of pro-inflammatory mediators, LD-associated proteins, and factors that control lipid metabolic pathways. These combined effects of IL-25 led to attenuated hepatic steatosis (Fig. S4). Conversely, IL-25 deficiency caused exacerbated hepatic lipid accumulation in HFD-fed mice. These results implicate IL-25 and the downstream type 2 cytokines as potential therapeutic agents for control of human hepatic steatosis and associated pathologies.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01-DK083418 (AZ), R01-AI/DK49316 (T.S-D), R01-DK061652 (HW), USDA CRIS project #8040-51000-058-00 (AS, JFU), and National Natural Science Fund of China No. 81460122 (AJW).
Abbreviations
- NAFLD
Non-alcoholic fatty liver disease
- ILC2
type 2 innate lymphoid cells
- HFD
high-fat diet
- NCD
normal control diet
- WT
wild type
- FA
fatty acid
- TG
triglyceride
- CIDE
cell death-inducing DFFA-like effector
- Hadh
hydroxyacyl-Coenzyme A dehydrogenase
- LD
lipid droplet
- M1
classically-activated
- M2
alternatively-activated
- PLIN
perilipin
Footnotes
Disclaimer: This article was prepared while Aiping Zhao was employed at the University of Maryland School of Medicine. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. The USDA is an equal opportunity provider and employer.
References
- 1.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic Fatty Liver Disease: Pathology and Pathogenesis. Annual Review of Pathology: Mechanisms of Disease. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 2.Jin C, Henao-Mejia J, Flavell R-á. Innate Immune Receptors: Key Regulators of Metabolic Disease Progression. Cell Metabolism. 2013;17:873–882. doi: 10.1016/j.cmet.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional Specialization of Interleukin-17 Family Members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Rickel EA, Siegel LA, Yoon BRP, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL. Identification of Functional Roles for Both IL-17RB and IL-17RA in Mediating IL-25-Induced Activities. The Journal of Immunology. 2008;181:4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- 5.Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cellular Signalling. 2011;23:1069–1075. doi: 10.1016/j.cellsig.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Jr, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, Bhandoola A, Artis D. IL25 elicits a multipotent progenitor cell population that promotes TH2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhao A, Urban JF, Sun R, Stiltz J, Morimoto M, Notari L, Madden KB, Yang Z, Grinchuk V, Ramalingam TR, Wynn TA, Shea-Donohue T. Critical Role of IL-25 in Nematode Infection-Induced Alterations in Intestinal Function. The Journal of Immunology. 2010;185:6921–6929. doi: 10.4049/jimmunol.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, Troy AE, Kobuley DE, Kastelein RA, Cua DJ, Yu Y, Artis D. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. The Journal of Experimental Medicine. 2008;205:2191–2198. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda K, Nakajima H, Suzuki K, Kagami S, Hirose K, Suto A, Saito Y, Iwamoto I. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood. 2003;101:3594–3596. doi: 10.1182/blood-2002-09-2817. [DOI] [PubMed] [Google Scholar]
- 12.Kang CM, Jang AS, Ahn MH, Shin JA, Kim JH, Choi YS, Rhim TY, Park CS. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am J Respir Cell Mol Biol. 2005;33:290–296. doi: 10.1165/rcmb.2005-0003OC. [DOI] [PubMed] [Google Scholar]
- 13.Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, Artis D. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarra M, Cupi ML, Bernardini R, Ronchetti G, Monteleone I, Ranalli M, Franze E, Rizzo A, Colantoni A, Caprioli F, Maggioni M, Gambacurta A, Mattei M, Macdonald TT, Pallone F, Monteleone G. IL-25 prevents and cures fulminant hepatitis through a myeloid-derived suppressor cell- dependent mechanism. Hepatology. 2013;58:1436–1450. doi: 10.1002/hep.26446. [DOI] [PubMed] [Google Scholar]
- 15.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPAR[gamma] signaling and metabolism: the good, the bad and the future. Nat Med. 2013;99:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, Grinchuk V, Smith A, Qin B, Bohl JA, Sun R, Notari L, Zhang Z, Sesaki H, Urban JF, Shea-Donohue T, Zhao A. Parasitic Nematode-Induced Modulation of Body Weight and Associated Metabolic Dysfunction in Mouse Models of Obesity. Infection and Immunity. 2013 doi: 10.1128/IAI.00053-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The Diagnosis and Management of Non-alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Zhao A, Yang Z, Sun R, Grinchuk V, Netzel-Arnett S, Anglin IE, Driesbaugh KH, Notari L, Bohl JA, Madden KB, Urban JF, Antalis TM, Shea-Donohue T. SerpinB2 Is Critical to Th2 Immunity against Enteric Nematode Infection. The Journal of Immunology. 2013;190:5779–5787. doi: 10.4049/jimmunol.1200293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, Habu Y, Miyazaki H, Hiroi S, Seki S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. Journal of Hepatology. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 21.Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, Nishiyama K, Yamamoto J, Seki S. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. Journal of Leukocyte Biology. 2013;94:1325–1336. doi: 10.1189/jlb.0313144. [DOI] [PubMed] [Google Scholar]
- 22.Price AE, Liang HE, Sulliva BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein IC. Kupffer cell heterogeneity: functional properties of bone marrow–derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, van Rooijen N, Staels B, Kersten S, Muller M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–522. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 25.Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, Sheridan R, Xanthakos SA, Steinbrecher KA, Sartor RB, Kohli R, Karp CL, Divanovic S. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830–1839. doi: 10.1002/hep.26746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu R, Tao A, Zhang S, Zhang M. Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochimica et Biophysica Sinica. 2013;45:726–733. doi: 10.1093/abbs/gmt065. [DOI] [PubMed] [Google Scholar]
- 27.Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, Han X, Peng Y, Chen X, Shen L, Qiu D, Li Z, Ma X. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clinical & Experimental Immunology. 2011;166:281–290. doi: 10.1111/j.1365-2249.2011.04471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okumura T. Role of lipid droplet proteins in liver steatosis. J Physiol Biochem. 2011;67:629–636. doi: 10.1007/s13105-011-0110-6. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Zhou L, Li P. CIDE Proteins and Lipid Metabolism. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:1094–1098. doi: 10.1161/ATVBAHA.111.241489. [DOI] [PubMed] [Google Scholar]
- 30.Zhou L, Xu L, Ye J, Li D, Wang W, Li X, Wu L, Wang H, Guan F, Li P. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology. 2012;56:95–107. doi: 10.1002/hep.25611. [DOI] [PubMed] [Google Scholar]
- 31.McManaman JL, Bales ES, Orlicky DJ, Jackman M, MacLean PS, Cain S, Crunk AE, Mansur A, Graham CE, Bowman TA, Greenberg AS. Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. Journal of Lipid Research. 2013;54:1346–1359. doi: 10.1194/jlr.M035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Comprehensive Physiology. John Wiley & Sons, Inc; 2013. Kupffer Cells in the Liver. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 34.Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. The FASEB Journal. 2011;25:4301–4311. doi: 10.1096/fj.11-189472. [DOI] [PubMed] [Google Scholar]
- 35.Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous Sp, Louvet A, Lafdil F, Pecker Fo, Tran A, Gual P, Mallat A, Lotersztajn S, Pavoine C. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130–142. doi: 10.1002/hep.26607. [DOI] [PubMed] [Google Scholar]
- 36.Chu D, Du M, Hu X, Wu Q, Shen J. Paeoniflorin attenuates schistosomiasis japonica-associated liver fibrosis through inhibiting alternative activation of macrophages. Parasitology. 2011;138:1258–1271. doi: 10.1017/S0031182011001065. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Gardner CR, Laskin JD, Laskin DL. Classical and alternative activation of rat hepatic sinusoidal endothelial cells by inflammatory stimuli. Exp Mol Pathol. 2013;94:160–167. doi: 10.1016/j.yexmp.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, Schopf L, Urban JF., Jr Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. 2004;201:139–155. doi: 10.1111/j.0105-2896.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 39.Akaiwa M, Yu B, Umeshita-Suyama R, Terada N, Suto H, Koga T, Arima K, Matsushita S, Saito H, Ogawa H, Furue M, Hamasaki N, Ohshima K, Izuhara K. Localization of human interleukin 13 receptor in non-haematopoietic cells. Cytokine. 2001;13:75–84. doi: 10.1006/cyto.2000.0814. [DOI] [PubMed] [Google Scholar]
- 40.Gabay C, Porter B, Guenette D, Billir B, Arend WP. Interleukin-4 (IL-4) and IL-13 enhance the effect of IL-1beta on production of IL-1 receptor antagonist by human primary hepatocytes and hepatoma HepG2 cells: differential effect on C-reactive protein production. Blood. 1999;93:1299–1307. [PubMed] [Google Scholar]
- 41.Stanya KJ, Jacobi D, Liu S, Bhargava P, Dai L, Gangl MR, Inouye K, Barlow JL, Ji Y, Mizgerd JP, Qi L, Shi H, McKenzie ANJ, Lee CH. Direct control of hepatic glucose production by interleukin-13 in mice. The Journal of Clinical Investigation. 2013;123:261–271. doi: 10.1172/JCI64941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie ANJ. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fina D, Franze E, Rovedatti L, Corazza GR, Biancone L, Sileri PP, Sica G, MacDonald TT, Pallone F, Di Sabatino A, Monteleone G. Interleukin-25 production is differently regulated by TNF-α and TGF-β1 in the human gut. Mucosal Immunology. 2011;4:239–244. doi: 10.1038/mi.2010.68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.