

Abstract
The last decade has witnessed significant growth in therapeutic options for patients diagnosed with lung cancer. This is due in major part to our improved technological ability to interrogate the genomics of cancer cells, which has enabled the development of biologically rational anticancer agents. The recognition that lung cancer is not a single disease entity dates back many decades to the histological subclassification of malignant neoplasms of the lung into subcategories of small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). While SCLC continues to be regarded as a single histologic and therapeutic category, the NSCLC subset has undergone additional subcategorizations with distinct management algorithms for specific histologic and molecular subtypes. The defining characteristics of these NSCLC subtypes have evolved into important tools for prognosis and for predicting the likelihood of benefit when patients are treated with anticancer agents.
Keywords: Biomarkers, lung cancer, NSCLC, EGFR, ALK, ROS1, RET
1.0 Introduction
The last decade has witnessed significant growth in therapeutic options for patients diagnosed with lung cancer. This is due in major part to our improved technological ability to interrogate the genomics of cancer cells, which has enabled the development of biologically rational anticancer agents. The recognition that lung cancer is not a single disease entity dates back many decades to the histological subclassification of malignant neoplasms of the lung into subcategories of small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). While SCLC continues to be regarded as a single histologic and therapeutic category, the NSCLC subset has undergone additional subcategorizations with distinct management algorithms for specific histologic and molecular subtypes. The defining characteristics of these NSCLC subtypes have evolved into important tools for prognosis and for predicting the likelihood of benefit when patients are treated with anticancer agents.
A discrete and measurable factor, whether in the whole patient or within the neoplastic cancer cells, that provides information on the likelihood of treatment efficacy is termed a predictive biomarker (1, 2). In contrast, a measurable factor that provides information on the overall patient outcome irrespective of treatment intervention is classically considered a prognostic biomarker (1, 2). Various biomarkers have emerged as predictive and prognostic markers in NSCLC patients and are now employed as part of their standard management. Putative biomarkers employed in clinical trials of investigational agents in SCLC, none of which have led to a management-defining paradigm, will be outside the scope of this review. This review will therefore focus on the clinical, histologic and molecular factors that are currently employed to guide the selection of therapeutic options for NSCLC patients.
2.0 Tumor histology as a biomarker in NSCLC
The WHO/IASLC classification of NSCLC includes various subtypes characterized by distinct morphology and immunophenotype (3, 4). The squamous and adenocarcinoma categories represent the two major histologic subtypes of NSCLC. The utility of tumor histology as a biomarker for selecting therapeutic intervention is therefore relevant to this review. The impact of squamous histology as a poor prognostic factor is supported by various retrospective and prospective studies (5, 6). This strategy became an established paradigm following retrospective analysis of outcome data from prospective studies of pemetrexed in unselected NSCLC patients, where a differential efficacy was noted between patients with squamous and non-squamous tumors (7, 8). Prospective comparison of the efficacy of pemetrexed-containing and gemcitabine-containing platinum doublet chemotherapy regimens as first line treatment of advanced NSCLC confirmed the differential efficacy of a pemetrexed-containing doublet by histology (9).
Histology has also served as a surrogate biomarker for increased risk of treatment-related toxicity leading to the avoidance of specific therapeutic agents. The notable example is the increased propensity for squamous tumors, which are more likely to be cavitary and centrally located in close proximity to major blood vessels, to hemorrhage following treatment with agents targeting angiogenesis such as bevacizumab (8). Squamous histology has thus become a biomarker to exclude patients who are unsuitable for anti-angiogenesis therapies. The main drawback with the use of tumor histology as a predictive biomarker in NSCLC is the significant discordance even among expert pulmonary pathologists in establishing a pathologic diagnosis of squamous NSCLC (10). Nonetheless, an algorithm that couples cell morphology and immunophenotype in the hands of an experienced pathologist can overcome this challenge in most cases.
3.0 Genetic alterations as biomarker
The major advance in the treatment of NSCLC in the last decade grew from the recognition that specific genetic alterations define subsets of NSCLC (11). This paved the way for the development of an array of effective agents to specifically counteract the biological consequences of such genetic aberrations. Thus, NSCLC went from a disease defined primarily by tumor histology to an amalgam of molecular subtypes, of which, the subsets characterized by alterations in the epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) genes are the most dominant.
3.1 Epidermal growth factor receptor (EGFR) gene mutations as a biomarker
The EGFR is a 170-kDa plasma membrane glycoprotein consisting of a large extracellular region, a single transmembrane domain, and an intracellular domain with tyrosine kinase activity and a C-terminal tail. The EGFR family consists of 4 closely related receptors, HER-1/ErbB1, HER-2/neu/ErbB2, HER-3/ErbB3 and HER-4/ErbB4 with significant homology in their kinase domains, but differences in the coding regions for the extracellular domain and the C-terminal tails (12). Dimerization of ErbB receptors upon ligand binding to the extracellular domain results in activation of their intrinsic tyrosine kinase activity. Activation of the EGFR receptor via phosphorylation relays downstream signals to the phosphatidylinositol 3-kinase (PI3K)/AKT and RAS/RAF/mitogen-activated protein kinase (MAPK) pathways. These intracellular signaling pathways that are responsible for the normal regulation of essential cellular processes such as proliferation and apoptosis are coopted by neoplastic cells harboring EGFR mutation (12, 13). Mutations in the EGFR gene occurring in NSCLC are commonly localized within the tyrosine kinase domain of the gene. Well established mutations include deletions in exon 19 (60%), missense mutation (L858R) in exon 21 (25%), point mutations in exons 18, 20 or 21, and insertion in exon 20 (14–16). These alterations result in constitutive activation of the kinase activity of EGFR and serve as the driver of neoplastic transformation and progression.
3.1.1 EGFR as a prognostic biomarker
Activating mutations in the EGFR gene confer a good prognosis on patients whose tumors harbor such alterations (17). In a prospective study that enrolled 647 patients for molecular profiling, 22.1% of NSCLC cases harbored a mutation in the EGFR gene. The patients with EGFR mutations had a much longer overall survival (OS, 3.51 years; 95% CI, 2.89 to 5.5 years), compared to patients without any detectable mutation (1.85 years; 95% CI, 1.61–2.13 years) (17). Patients with exon 19 deletion type EGFR mutation have also been shown to have significantly longer OS compared to patients with L858R mutation. A retrospective study of 32 patients showed an increased OS (38 vs 17 months) in patients with EGFR exon 19 deletion compared to patients with L858R mutation (18). Similar observation was made by Riely et al who demonstrated a significantly longer OS (34 vs 8 months) in patients with exon 19 deletions over those with L858R substitution (19). The prognostic impact of the less common types of EGFR mutations involving exon 18 or 20 has not been well studied due to the lower prevalence of these genetic alterations.
3.1.2 Predicting efficacy of EGFR inhibitors
Activating mutations in the gene encoding the EGFR protein are present in approximately 10–20% of NSCLC patients diagnosed in North America and Western Europe, and 30–50% of Asian patients (20). These mutations are commonly found within defined hotspots in the gene namely, exon 18, 19, 20 and 21 (14–16). In addition to its recognized role as a prognostic biomarker, EGFR mutation also reliably predicts the efficacy of EGFR targeted agents in prospective studies comparing cytotoxic chemotherapy to biologic agents targeting the activated kinase function of mutant EGFR. Consistent with the better prognosis associated with exon 19 deletion mutation, EGFR inhibitor therapy is also associated with superior efficacy in patients with exon 19 deletion compared to those with exon 21 alterations. It is noteworthy that the performance of different EGFR mutations as a predictive biomarker of treatment efficacy varies. For instance, while exon 19 and 21 alterations are generally sensitizing, rarer alterations involving exon 18 and 20 may or may not be. Particularly, exon 20 insertion, which is a rare subtype at 4% of all EGFR mutations but represents the third most common EGFR mutation, has approximately 122 different variants most of which are not responsive to currently available kinase inhibitors (21). A retrospective study of 23 Korean patients with exon 20 insertion reported objective response in only four out of 16 (25%) patients treated with gefitinib. Median OS of the responding patients was 23 months vs 7 months for the non-responders. Eight of the 16 (50%) treated patients had concurrent mutations including alterations in exons 21 and 18. Of note, different exon 20 mutations and other coexisting mutations appeared to have a different role on treatment response (22). Another retrospective study identified 27 patients with exon 20 insertion and the most common variant described was (V769_D770insASV), accounting for 22%. The median OS was 16 months (20). Thus, patients with exon 20 insertion have survival rates similar to those seen in EGFR wild-type NSCLC. These patients may not be best suited for currently available EGFR targeted agents. An intriguing response to HSP 90 inhibitor was reported in preclinical studies (23) as well as in early phase clinical study, leading to an ongoing prospective evaluation of AUY922 as treatment for NSCLC patients whose tumors harbor EGFR exon 20 mutations.
To date, many clinical trials have been conducted using EGFR as a predictive biomarker. Overexpression of EGFR protein whether assessed by in situ hybridization or immunohistochemistry failed to reliably identify patients likely to benefit from biologic agents targeted against the kinase activity of EGFR receptor. Activating mutation in the kinase domain of the EGFR gene is the most reliable predictive biomarker for this class of agents and has been successfully employed in paradigm-defining clinical trials of EGFR inhibitors in lung cancer. Please refer to the comprehensive review of EGFR targeted therapies by Conor et al in this issue of the journal.
3.1.3 Predicting resistance to EGFR tyrosine kinase inhibitors
Loss of treatment efficacy is nearly universal in patients treated with EGFR targeted agents (24). Various mechanisms that mediate resistance have been uncovered and can be classified as pharmacologically or biologically mediated resistance (Table 1). Pharmacological resistance is an acquired resistance that occurs through inadequate drug delivery to the target due to various factors such as limited delivery to sanctuary sites like the central nervous system, poor adherence to dosing schedule, decreased gastrointestinal absorption, and altered hepatic metabolism. The fluctuations and low levels of drug exposure consequently facilitate the development of resistance by cancer cells exposed to suboptimal drug concentration.
Table 1.
Mechanisms of acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC
| Pharmacological mechanisms | Inadequate CNS penetration Non-compliance/dose reduction or interruption Smoking Reduced absorption Increased hepatic metabolism |
|
| Biological mechanisms | Altered drug target ~ 60% (24) | T790M alone ~40–60% (32, 127) T790M with EGFR amplification ~10% (127) Other EGFR mutations ~1–2% (127) |
| Bypass tracks ~ 20% (24) | MET amplification ~5% (32, 127) HER2 amplification ~ 8–13% (32) PIK3CA ~1–2% (128) |
|
| Altered Phenotype | EMT ~1–2% (32, 127, 129) SCLC ~6% (32, 127) |
|
| Unknown mechanism | Accounts for 18–20% of acquired resistance (32, 127) | |
Biological mechanisms of resistance can develop through a positive selection pressure that favors the outgrowth of specific subpopulations of cancer cells that are able to adapt and proliferate in the presence of the EGFR inhibitor. Adaptations in the resistant clones may occur either through an alteration in the target or by activation of alternative or bypass signaling pathways (24). Alterations in the drug target preserve the oncogenic drive despite adequate drug exposure. The T790 mutation acquired following extended period of therapy with an EGFR inhibitor is a classic example of this type of resistance mechanism. This mutation is the most frequently described and may be found in more than 50% of patients with acquired resistance to an EGFR tyrosine kinase inhibitor (25). Similar but rarer mutations associated with resistance include D761Y and L747S mutations (26, 27). Activation of bypass signaling pathways is the other common mechanism of biological resistance observed in EGFR-mutant patients. In this scenario, an alternative receptor tyrosine kinase is coopted to reactivate a critical signaling cascade downstream of the EGFR kinase blockade. Escape from the consequence of target inhibition enables the cell to continue to proliferate and survive in the face of an effective drug level and preserved drug target. MET gene amplification is the most commonly observed bypass signaling pathway that mediates failure of EGFR inhibitors (28, 29), but mutations in the PIK3CA and BRAF genes as well as HER2 gene amplification can have the same consequence (30–32).
3.2 Anaplastic lymphoma kinase (ALK)
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase whose biological function is yet to be defined but is normally expressed in the small intestine, testes and the nervous system of adult human tissues (33). Morris and colleagues first reported oncogenic activity of ALK in anaplastic large cell lymphoma more than 2 decades ago (33). In 2007, Soda and colleagues in Japan used retrovirus-mediated expression screening to identify a novel rearrangement of the ALK gene in NSCLC. The genes encoding echinoderm microtubule associated protein like 4 (EML4) and ALK are both located on the short arm of chromosome 2 (2p21 and 2p23) (34). An inversion mutation within the short arm of chromosome 2 (p21p23) results in the formation of a fusion gene comprising EML4 and ALK, whereby exon 1–13 of EML4 is joined to exon 20–29 of ALK. The EML4-ALK fusion kinase is potently oncogenic and is the driver in a subgroup of NSCLC patients (34). The coiled coil domain of EML4-ALK is involved in cellular proliferation and apoptosis inhibition through downstream signaling relays involving PI3K/AKT, MAPK/extracellular-related kinase (ERK1/2) and JAK/STAT pathways (35, 36). Since the initial discovery of the ALK-EML4 fusion protein, other fusion partners of ALK have been identified (35). To date, 11 different ALK fusion variants have been reported based on demonstrable oncogenic activity in NIH-3T3 cells or in Ba/F3 cells (35, 37). These include fusions of ALK with tropomyosin-related kinase (TRK), kinesin family member 5B (KIF5B), kinesin light chain 1 (KLC1), protein tyrosine phosphatase, nonreceptor type 3, huntingtin interacting protein 1 (HIP1) and TRP (38–42).
The ALK-EML4 fusion oncogene is reported in 2–7% of advanced NSCLC tumors (43). Initial reports from Soda et al showed five (6.7%) out of 75 Japanese NSCLC patients were positive for EML4–ALK (34). Other studies showed variable frequency of ALK fusion in lung cancer. Taheuchi et al reported a frequency of 4.4% (11 of 253) in Japanese patients (44). A lower frequency of 1.4% was reported in 136 samples obtained from Caucasian patients while a rate of 3.6% was reported in Korean patients (45). Similarly, a rate of 4.9% was described in 266 Chinese patients tested for EML4-ALK (46), while a prevalence of 7.5% was reported in a population of Italian and Spanish patients (47). Using clinical characteristic such as female gender, Asian ethnicity, never or light smoker and adenocarcinoma histology, Shaw et al from the Massachusetts General Hospital screened 141 tumor samples of which 19 (13%) cases were found to harbor EML4-ALK translocation (48). Overall, EML4-ALK gene translocation more frequently presents in patients who are relatively young, male, with light or no smoking history, and predominantly with adenocarcinoma histology (34, 35, 37, 48). Nonetheless, these clinical characteristics are relatively insensitive and imprecise as a predictive biomarker to select patients for ALK-targeted therapies. Indeed, the first EML4-ALK fusion gene was identified in a smoker with lung cancer.
3.2.1 ALK translocation as a prognostic biomarker
Patients whose tumors harbor alterations in the ALK gene appear to have a worse prognosis than those without ALK alteration. In an observational study in non-smoking patients with advanced adenocarcinoma, patients with ALK positive disease had a higher risk of disease progression in comparison to ALK negative patients (49). ALK translocation has also been associated with an increased risk of brain involvement (40% vs 21%), liver metastases and a greater number of metastatic sites (49, 50). Kim et al reported a shorter median OS of 14.3 vs 33.3 months in patients with ALK(+) vs ALK(−) disease who were well matched with respect to age, gender, stage and smoking status (51). Similar observations were made by other investigators, although there have been occasional reports that failed to show ALK translocation as a negative prognostic biomarker. While the majority of patients with known diagnosis of ALK(+) NSCLC presents at advanced stages (52), the prognostic role of ALK translocation has also been studied in early stage disease. Paik et al analyzed 735 cases of stage I–III NSCLC and found 3.8% of cases positive for ALK translocation. There was no significant difference in the mean OS (97.7 vs 78.9 months; p=0.10) and disease free survival (76.4 vs 71.3 months; p = 0.66) between ALK(+) and ALK(−) patients (53). However, ALK rearranged tumors had a lower stage of the primary site but more frequent regional lymph node involvement (53–56).
3.2.2 ALK translocation as a predictive biomarker
ALK gene translocation assessed by FISH assay has been used successfully as a predictive biomarker of the efficacy of different agents targeting the ALK kinase activity including crizotinib, ceritinib and alectinib. This has resulted in the approval of two biologic agents, crizotinib and ceritinib, by the US FDA as standard therapy for the ALK(+) subset of NSCLC. Crizotinib is a first in class, orally available small molecule with potent inhibitory effects on cell proliferation through the induction of apoptosis and arrest in G1-S phase cell-cycle (57). The ALK kinase inhibition induced by crizotinib results in potent suppression of downstream survival signaling and induction of apoptosis (58). Initial phase 1 clinical trial experience in previously treated NSCLC patients harboring ALK translocation showed an objective response rate (ORR) of 60.8% and median progression free survival (PFS) of 9.7 months, leading to FDA approval of crizotinib in this subset of NSCLC (43, 59). Subsequently, randomized comparison of crizotinib to standard second line chemotherapy (docetaxel or pemetrexed) in 347 patients previously treated with platinum-based chemotherapy confirmed the superiority of crizotinib with a median PFS of 7.7 months vs 3 months and response rates of 65% vs 20% (60). More recently, the benefit of crizotinib over frontline platinum-based doublet chemotherapy (pemetrexed plus cisplatin or carboplatin) in newly diagnosed patients with advanced ALK-positive non-squamous NSCLC was demonstrated in a randomized phase III study showing a PFS of 10.9 vs 7 months and response rate of 74% vs 45% with crizotinib and chemotherapy, respectively (61).
Newer ALK inhibitors such as ceritinib, alectinib and brigatinib (AP26113) with greater potency over crizotinib have been evaluated in the clinical setting. Ceritinib, which is 5 to 20 times more potent than crizotinib, has a distinct chemical structure, is orally available, and has activity against some of the known mechanisms mediating resistance to crizotinib. It, however, lacks activity against the G1202R and F1174C mutations in preclinical models (62–65). Ceritinib achieved a response rate of 58% and a median PFS of 7 months when tested in 130 patients with ALK positive NSCLC. This efficacy was independent of the type of ALK gene resistance mutations consistent with the in vitro modeling experiments (66). This study led to the approval of ceritinib as a standard treatment for patients with ALK translocated NSCLC who have failed crizotinib (67). Other new generation ALK kinase inhibitors such as alectinib with potent in vitro activity have also shown interesting clinical activity at extracranial and intracranial sites (68, 69).
3.2.3 ALK-fusion partners and EML4 variants as predictors of efficacy
Following the original description of the ALK and EML4 fusion, other fusion partners have been identified including TRK, KIF5B, KLC1, HIP1 and TRP (38–42). The currently approved diagnostic testing for ALK translocation positive lung cancer is the Vysis break-apart FISH assay, which does not differentiate between various fusion partners of ALK. It is therefore currently unknown whether or not the partner protein with ALK influences response of patients to targeted therapies. It is however, noteworthy that similar to the experience with EML4-ALK translocation, crizotinib has been shown to achieve comparable efficacy in patients harboring novel fusion partner translocations (41).
Similarly, the breakpoint region in the EML4 gene can vary, resulting in significant differences in the physicochemical characteristics of the EML4-ALK fusion protein variants. To date, up to 11 different EML4-ALK variants have been described and studies of the biological function of the protein variants have been conducted with intriguing results (70). Wu et al compared EML4-ALK fusion variants in 39 patients including 24 with variant 1 and 15 with non-variant 1 fusion genes (two v2, six v3a, five v3b, and two other variant types). There was no difference in age, sex or OS (14.1 vs 16.8 months; p=0.869) for variant 1 and non-variant 1 cases but the variant 1 cases were more likely to be heavy smokers (54). In preclinical work using Ba/F3 cell lines to test the efficacy of ALK kinase and HSP90 inhibitors against different fusion variants of EML4-ALK, v2, which is the least stable protein variant, showed the greatest sensitivity to ALK kinase inhibition, v1 and v3b showed intermediate sensitivity, while the v3a variant was the least sensitive. The findings from these preclinical experiments suggest that ALK fusion variants with different physicochemical properties may also differ in terms of drug sensitivity and responsiveness to ALK kinase inhibitors (71). The overall rarity of ALK translocated NSCLC and the even rarer frequency of subsets defined by the different fusion protein variants make clinical confirmation of this observation very challenging. Notably, correlative analysis using banked tissue samples collected as part of prospective studies of crizotinib did not show any significant association between mutant variants and response to therapy (43). It is anticipated that larger numbers of banked tissue samples from ongoing and future prospective clinical trials could be harnessed to elucidate the impact of fusion partner proteins and fusion protein variants as predictive biomarkers of treatment efficacy.
3.2.4 Alterations in ALK and other genes predicting resistance to ALK inhibitors
Despite the impressive efficacy of targeted agents such as crizotinib and ceritinib in ALK-translocated lung cancer, the vast majority of patients ultimately progress within a median period of 9–13 months. The mechanisms responsible for treatment failure have been elucidated using preclinical models as well as through detailed analysis of tissue samples obtained from patients at the time of progression (72–74). Resistance to ALK targeted therapy is now known to occur either in an ALK-dependent manner, whereby the cancer cells remain dependent on ALK signaling, or through an ALK-independent process (Figure 1). Acquired mutations in the ALK kinase domain as well as copy number gains of the fusion gene can impair the ability of the targeted therapy to inhibit the fusion protein. Such acquired mutations include L1196M, S1206Y, C1156Y, G1202R, 1151Tins and L1152R occurring around the ATP binding site of the kinase enzyme, which lead to a restoration of ALK signaling in the presence of crizotinib (72, 74–76). ALK-independent mechanisms of resistance include the cooption by the cancer cells of alternative oncogenic drivers, such as KRAS and EGFR, or through ligand-driven activation of the HER family, IGF-1R and KIT (74, 76–80). It has yet to be shown whether these bypass mechanisms occur de novo in previously untreated patients. Moreover, these genetic alterations appear to be agent-specific since new generation inhibitors of ALK fusion protein, such as alectinib and ceritinib, demonstrated efficacy in the salvage setting across patient subgroups with different mechanisms of resistance to crizotinib (66). Nonetheless, novel mutations, such as the V1180L gatekeeper mutation and the I1171T mutations occurring post alectinib, remain sensitive to ceritinib (81–83). It is conceivable that these resistance mutations will guide the choice of salvage therapy in patients who have failed a prior ALK-targeted agent.
Figure 1.
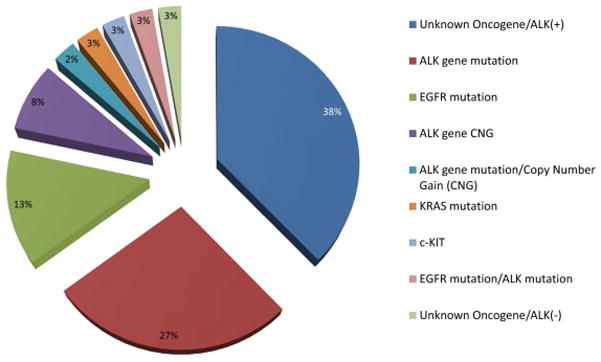
Mechanisms of resistance to ALK inhibitor therapy reported in published data generated from tumor samples obtained from patients with acquired resistance to crizotinib (74, 78, 84)
3.3 KRAS mutations
KRAS is the most commonly detected mutation in NSCLC, present in up to a third of all cases (85). It is more common in tumors with adenocarcinoma histology as opposed to squamous type NSCLC (86). Although there is currently no targeted therapy with established efficacy in NSCLC harboring mutant KRAS, this genetic mutation was previously considered a negative predictive biomarker for efficacy of EGFR targeted inhibitors. However, this presumed negative association has not been conclusively borne out by larger studies (87, 88). A meta-analysis of 17 studies reporting on a total of 165 patients with tumors harboring KRAS mutation suggested that concurrent presence of KRAS and EGFR mutations showed a significant association with lack of clinical benefit from EGFR inhibitors (89). Mao et al also conducted a separate meta-analysis of 22 studies and 231 patients with KRAS mutation. They reported an ORR of 3% vs 26% in patients whose tumors harbor mutant EGFR and mutant or wild type KRAS gene, respectively. The overall pooled relative rate of response in the presence of KRAS mutation was reported as 0.29 (95% CI: 0.18–0.47; P<0.01). There was, however, no significant difference in OS (90). The presence of KRAS mutation was previously considered a marker of poor prognosis based on small data series reported from single institution studies (91–93). Recently, Riely et al conducted a comprehensive assessment in 677 patients with advanced recurrent NSCLC looking at the frequency and impact of KRAS mutation as well as the impact of specific codon mutation, and failed to show any association with outcome (94). The study showed that KRAS mutation, whether codon 12 or 13, had no significant impact on survival. Also, comparison of KRAS transition type mutation with transversion type mutations also showed no significant difference in outcome (p = 0.99). While patients whose tumors harbor KRAS codon 13 mutation had shorter OS compared with patients with codon 12 mutation (1.1 vs 1.3 years; p = 0.009), this finding could not be confirmed in an independent validation set consisting of samples from 682 patients with KRAS mutant lung cancers (1.0 vs 1.1 years, respectively; p=0.41) (94). Nonetheless, KRAS mutation remains the most frequently detected genetic alteration in NSCLC. While it does not at present offer any clinical value either as a prognostic indicator or as a therapeutic guide, targeted therapies against activating KRAS mutation are undergoing active testing as a therapeutic strategy in lung cancer. Table 2 provides a summary of ongoing clinical trials in KRAS mutant NSCLC. The success of some of these clinical trials may establish KRAS as a bona fide predictive biomarker in NSCLC.
Table 2.
Active clinical trials of therapies targeting KRAS mutant lung cancer
| Study | Treatment | Design | Primary Endpoint | Rationale and Strategy |
|---|---|---|---|---|
| SELECT-1 study; (NCT01933932) | Selumetinib + Docetaxel | Randomized phase III | PFS | Improved efficacy of the combination over docetaxel alone |
| Phase II Study of VS-6063 (NCT01951690) | VS-6063 (Defactinib) | Phase II | PFS | Demonstrate if VS-6063 improves PFS within each cohort |
| AZD6244 in Combination With Docetaxel, Compared With Docetaxel Alone, (NCT00890825) | AZD6244 + Docetaxel | Randomized, Phase II | OS | Compare the efficacy of the combination over docetaxel alone |
| Phase II Study of AZD6244 MEK-Inhibitor With Erlotinib (NCT01229150) | AZD6244 + Erlotinib | Randomized, Phase II | PFS | Compare the efficacy of combination over erlotinib alone |
| Erlotinib Plus Tivantinib (ARQ197) Versus Single Agent Chemotherapy (NCT01395758) | RQ 197 + erlotinib Vs. Pemetrexed, Docetaxel or Gemcitabine | Randomized, Phase II | PFS | Compare efficacy of combination erlotinib plus tivantinib over single agent chemotherapy. |
| Phase I/IB Trial of MEK162 in Combination With Erlotinib (NCT01859026) | MEK162 + Erlotinib | Non-Randomized, Phase I/IB | Maximum Tolerated Dose (MTD) | Safety of the combination MEK162 and erlotinib |
| Phase I/II Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination With the MEK Inhibitor PD-0325901 (NCT02022982) | Palbociclib + PD-0325901 | Phase I/II | Safety and Maximum Tolerated Dose (MTD) | Evaluate safety and compare efficacy of palbociclib in combination with another experimental drug PD-0325901 |
| Phase 1 Study of Trametinib in Combination With Chemoradiation (NCT01912625) | Trametinib + Chemoradiation | Phase I | DLT | Evaluate safety of trametinib in combination with chemotherapy and radiation therapy |
| JUNIPER (NCT02152631) | Abemaciclib + Best Supportive Care Vs. Erlotinib Plus Best Supportive | Randomized Phase III | PFS OS |
Compare efficacy of abemaciclib over erlotinib |
3.4 Rare genetic alterations as biomarkers
In addition to EGFR and ALK gene alterations which can be present in up to 40% of NSCLC patients, other genetic alterations involving ROS1, MET, RET, BRAF, and HER2, among others, have been described in smaller subsets of NSCLC. These alterations are currently employed as predictive biomarkers for therapeutic agents likely to be effective in patient subsets defined by the presence of these mutations, based on supporting clinical experience in other tumor types and/or from preclinical models.
3.4.1 ROS1
ROS1 was initially discovered as homolog of chicken c-ros, encodes a receptor tyrosine kinase and has significant homology with ALK kinase (95). It is arranged as an intracellular C-terminal tyrosine kinase domain and a large extracellular N-terminal domain. The normal biologic function of ROS1 has not yet been defined but it is highly expressed in the kidney but not in the lung (95, 96). The initial identification of ROS1 fusion gene as a driver of NSCLC arose from preclinical and correlative work conducted in the HCC78 cell line and in a patient tumor sample where SLC34A2 and CD74 were observed to be fused to the transmembrane region of ROS, resulting in a constitutively active truncated fusion protein with two transmembrane domains (39). Subsequently, Rimkunas and group reported 9 (1.6%) tumors expressing ROS1 in Chinese NSCLC patients with CD74-ROS1, SLC34A2-ROS1, and FIG-ROS1 fusions determined by reverse transcriptase PCR (97). Multiple ROS1 fusion proteins have been described and include SDC4, EZR, SLC34A2, TPM3, LIMA1 (LIM domain and actin binding 1), LRIG3 and MSN (98–101). Analysis of archival tumor samples revealed a prevalence of 1.7 to 2.4% with most patients being relatively young non-smokers with tumors of adenocarcinoma histology (102, 103).
The presence of ROS1 translocation has not been associated with prognostic difference in early stage lung cancer but a negative impact was noted in patients with advanced stage tumors harboring ROS1 relative to EGFR mutant patients (103). There is partial homology between ROS1 and ALK and preclinical studies have shown activity of crizotinib against a ROS1(+) cell line (102). Clinical evaluation of crizotinib in 50 patients with advanced NSCLC harboring ROS1 translocation showed an ORR of 72%, including three patients with complete response with median duration of response of 17.6 months and median PFS of 19.2 months (98). The most common ROS1 fusion partner is CD74 (44%) but there was no significant difference in the efficacy of crizotinib against the CD74 fusion partner as compared to other fusion partners (98). As expected, resistance to crizotinib has been observed in patients with ROS1(+) NSCLC. Awad et al reported the experience with a patient whose tumor harbored the CD74–ROS1 rearrangement treated with crizotinib. Initial response followed by resistance led to the identification of an acquired mutation with a glycine to arginine substitution at codon 2032 (G2032R) in the ROS1 kinase domain (104). Newer agents such as foretinib (GSK1363089), which is more potent than crizotinib and also has activity against the acquired G2032R mutation that mediates crizotinib resistance, have already been identified and are now in clinical development (100).
3.4.2 HER2
HER2, human epithelial receptor 2 (HER2/erbB2) is a member of the HER family that is activated by homo-or heterodimerization with another member of the erbB family (erbB1-4) leading to downstream activation of the PI3K/AKT/mammalian target of rapamycin (mTOR) pathway (105, 106). Amplification of the gene encoding HER2 and/or protein overexpression is well established as a biomarker for HER2 targeted therapies in breast, ovarian, gastric and uterine cancers (107–109). Mutation involving the tyrosine kinase domain of the erbB2 gene has also been reported in NSCLC at a prevalence of 1.6% and with a predilection for never smokers and adenocarcinoma histology but without regard to sex, race, or tumor stage (105, 109–111). A meta-analysis of published data showed HER2 protein expression but not HER2 gene amplification to be a marker of poor prognosis in lung cancer (112). Trastuzumab, a humanized monoclonal antibody against HER2, showed antitumor activity in preclinical models of NSCLC both alone and in combination with cytotoxic agents (113). However, HER2 protein expression failed to predict patients likely to benefit from trastuzumab or lapatinib, a tyrosine kinase inhibitor of HER1 and HER2, in clinical trials of patients with HER2 deregulated NSCLC (114, 115). There are multiple clinical trials currently underway examining newer generation kinase inhibitors such as dacomitinib, afatinib, and neratinib for HER2(+) lung cancer.
3.4.3 RET
Rearranged in transfection (RET) is a receptor tyrosine kinase that has been established as a driver mutation in various cancers including medullary thyroid cancer and subset of papillary thyroid cancer (11, 116, 117). RET oncogene aberration in lung cancer was first reported in a young, male, never smoker with metastatic NSCLC. Using parallel whole-genome and transcriptome sequencing, a fusion gene between KIF5B and the RET proto-oncogene caused by a pericentric inversion of 10p11.22–q11.21 was identified (118). Subsequent screening of larger sets of NSCLC tumor samples found RET fusions in approximately 1–2% of NSCLC, almost exclusively in adenocarcinoma (101, 117, 119–122). Although KIF5B is the most common fusion partner with RET, being present in 90% of reported cases, other fusion partners have been described including CCDC6, NCOA4, and TRIM33 (117, 119, 123, 124). Preclinical data demonstrated the activity of RET inhibitors in lung cancer cell lines harboring activating RET fusions (119, 122–124). The initial evidence for clinical efficacy of these agents in patients is mostly anecdotal, albeit with positive and encouraging results (125). Cabozantinib, an inhibitor of multiple kinases including RET, is currently being studied in a prospective phase II trial. Experience with the first three patients with RET fusion-positive NSCLC enrolled on the study was reported by Drilon et al, with two of the three patients achieving a partial response and the third patient experiencing stable disease (126). Currently, multiple clinical trials are underway using the presence of RET fusion as a predictive biomarker to select NSCLC patients for prospective evaluation of the clinical efficacy of RET inhibitors such as cabozantinib (NCT01639508), lenvatinib (NCT01877083), ponatinib (NCT01813734) and vandetanib (NCT01823068).
4.0. Conclusions
NSCLC has evolved into a conglomerate of tumor subgroups characterized by specific molecular aberrations rather than by simple origination from the lung. Most of the genetic alterations described to date present valid targets for therapeutic intervention with varying success, as demonstrated by the FDA approval of agents targeting EGFR and ALK alterations. Other rarer mutations such as ROS1 and RET have also been successfully targeted whereas attempts to target RAS gene alterations remain a work in progress. While these genetic alterations meet the basic definition of predictive biomarkers, their prognostic value has not been well characterized. Challenges related to sequence of testing, i.e. single assay vs multiplex assay, reflex testing vs testing on request, assay performance, and the comparison of platforms for detecting genetic alterations between immunohistochemistry, FISH, targeted DNA sequencing and next generation sequencing assay, continue to evolve as technological capabilities advance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Biomarkers Definitions Working G. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clinical pharmacology and therapeutics. 2001;69(3):89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 2.Oldenhuis CN, Oosting SF, Gietema JA, de Vries EG. Prognostic versus predictive value of biomarkers in oncology. European journal of cancer. 2008;44(7):946–53. doi: 10.1016/j.ejca.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. The European respiratory journal. 2001;18(6):1059–68. doi: 10.1183/09031936.01.00275301. [DOI] [PubMed] [Google Scholar]
- 4.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–85. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsch FR, Spreafico A, Novello S, Wood MD, Simms L, Papotti M. The prognostic and predictive role of histology in advanced non-small cell lung cancer: a literature review. J Thorac Oncol. 2008;3(12):1468–81. doi: 10.1097/JTO.0b013e318189f551. [DOI] [PubMed] [Google Scholar]
- 6.Clark GM. Prognostic factors versus predictive factors: Examples from a clinical trial of erlotinib. Molecular oncology. 2008;1(4):406–12. doi: 10.1016/j.molonc.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scagliotti G, Brodowicz T, Shepherd FA, Zielinski C, Vansteenkiste J, Manegold C, et al. Treatment-by-histology interaction analyses in three phase III trials show superiority of pemetrexed in nonsquamous non-small cell lung cancer. J Thorac Oncol. 2011;6(1):64–70. doi: 10.1097/JTO.0b013e3181f7c6d4. [DOI] [PubMed] [Google Scholar]
- 8.Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(36):5311–20. doi: 10.1200/JCO.2010.28.8126. [DOI] [PubMed] [Google Scholar]
- 9.Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–51. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 10.Grilley-Olson JE, Hayes DN, Moore DT, Leslie KO, Wilkerson MD, Qaqish BF, et al. Validation of interobserver agreement in lung cancer assessment: hematoxylin-eosin diagnostic reproducibility for non-small cell lung cancer: the 2004 World Health Organization classification and therapeutically relevant subsets. Archives of pathology & laboratory medicine. 2013;137(1):32–40. doi: 10.5858/arpa.2012-0033-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berge EM, Doebele RC. Targeted therapies in non-small cell lung cancer: emerging oncogene targets following the success of epidermal growth factor receptor. Seminars in oncology. 2014;41(1):110–25. doi: 10.1053/j.seminoncol.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacology & therapeutics. 2004;102(1):37–46. doi: 10.1016/j.pharmthera.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. Epub 2004/05/01. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Chavez A, Thomas A, Rajan A, Raffeld M, Morrow B, Kelly R, et al. Molecular Profiling and Targeted Therapy for Advanced Thoracic Malignancies: A Biomarker-Derived, Multiarm, Multihistology Phase II Basket Trial. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.58.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(14 Pt 2):4416s–20s. doi: 10.1158/1078-0432.CCR-06-0555. [DOI] [PubMed] [Google Scholar]
- 19.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(3 Pt 1):839–44. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 20.Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013;8(2):179–84. doi: 10.1097/JTO.0b013e3182779d18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. The Lancet Oncology. 2012;13(1):e23–31. doi: 10.1016/S1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 22.Wu JY, Wu SG, Yang CH, Gow CH, Chang YL, Yu CJ, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14(15):4877–82. doi: 10.1158/1078-0432.CCR-07-5123. [DOI] [PubMed] [Google Scholar]
- 23.Xu W, Soga S, Beebe K, Lee MJ, Kim YS, Trepel J, et al. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. British journal of cancer. 2007;97(6):741–4. doi: 10.1038/sj.bjc.6603950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nature reviews Clinical oncology. 2014;11(8):473–81. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 25.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. Epub 2005/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(21):6494–501. doi: 10.1158/1078-0432.CCR-06-1570. Epub 2006/11/07. [DOI] [PubMed] [Google Scholar]
- 27.Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26(7):1182–4. doi: 10.1200/JCO.2007.14.9039. author reply 4–6. Epub 2008/03/04. [DOI] [PubMed] [Google Scholar]
- 28.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43. doi: 10.1126/science.1141478. Epub 2007/04/28. [DOI] [PubMed] [Google Scholar]
- 29.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–7. doi: 10.1073/pnas.0710370104. Epub 2007/12/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–33. doi: 10.1073/pnas.1203530109. Epub 2012/07/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer discovery. 2012;2(10):922–33. doi: 10.1158/2159-8290.CD-12-0108. Epub 2012/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(8):2240–7. doi: 10.1158/1078-0432.CCR-12-2246. Epub 2013/03/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 34.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. Epub 2007/07/13. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Zhang S, Yang X, Yang J, Zhou Q, Yin L, et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Molecular cancer. 2010;9:188. doi: 10.1186/1476-4598-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer science. 2008;99(12):2349–55. doi: 10.1111/j.1349-7006.2008.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. European journal of cancer. 2010;46(10):1773–80. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R, et al. KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012;7(2):e31323. doi: 10.1371/journal.pone.0031323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 40.Choi YL, Lira ME, Hong M, Kim RN, Choi SJ, Song JY, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol. 2014;9(4):563–6. doi: 10.1097/JTO.0000000000000093. [DOI] [PubMed] [Google Scholar]
- 41.Fang DD, Zhang B, Gu Q, Lira M, Xu Q, Sun H, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol. 2014;9(3):285–94. doi: 10.1097/JTO.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 42.Hong M, Kim RN, Song JY, Choi SJ, Oh E, Lira ME, et al. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. J Thorac Oncol. 2014;9(3):419–22. doi: 10.1097/JTO.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 43.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14(20):6618–24. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 45.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14(13):4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115(8):1723–33. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- 47.Martelli MP, Sozzi G, Hernandez L, Pettirossi V, Navarro A, Conte D, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174(2):661–70. doi: 10.2353/ajpath.2009.080755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(8):2081–6. doi: 10.1158/1078-0432.CCR-10-1591. [DOI] [PubMed] [Google Scholar]
- 49.Yang P, Kulig K, Boland JM, Erickson-Johnson MR, Oliveira AM, Wampfler J, et al. Worse disease-free survival in never-smokers with ALK+ lung adenocarcinoma. J Thorac Oncol. 2012;7(1):90–7. doi: 10.1097/JTO.0b013e31823c5c32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fallet V, Cadranel J, Doubre H, Toper C, Monnet I, Chinet T, et al. Prospective screening for ALK: clinical features and outcome according to ALK status. European journal of cancer. 2014;50(7):1239–46. doi: 10.1016/j.ejca.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Kim HR, Shim HS, Chung JH, Lee YJ, Hong YK, Rha SY, et al. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 2012;118(3):729–39. doi: 10.1002/cncr.26311. [DOI] [PubMed] [Google Scholar]
- 52.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paik JH, Choi CM, Kim H, Jang SJ, Choe G, Kim DK, et al. Clinicopathologic implication of ALK rearrangement in surgically resected lung cancer: a proposal of diagnostic algorithm for ALK-rearranged adenocarcinoma. Lung cancer. 2012;76(3):403–9. doi: 10.1016/j.lungcan.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 54.Wu SG, Kuo YW, Chang YL, Shih JY, Chen YH, Tsai MF, et al. EML4-ALK translocation predicts better outcome in lung adenocarcinoma patients with wild-type EGFR. J Thorac Oncol. 2012;7(1):98–104. doi: 10.1097/JTO.0b013e3182370e30. [DOI] [PubMed] [Google Scholar]
- 55.Lee JK, Park HS, Kim DW, Kulig K, Kim TM, Lee SH, et al. Comparative analyses of overall survival in patients with anaplastic lymphoma kinase-positive and matched wild-type advanced nonsmall cell lung cancer. Cancer. 2012;118(14):3579–86. doi: 10.1002/cncr.26668. [DOI] [PubMed] [Google Scholar]
- 56.Zhang NN, Liu YT, Ma L, Wang L, Hao XZ, Yuan Z, et al. The molecular detection and clinical significance of ALK rearrangement in selected advanced non-small cell lung cancer: ALK expression provides insights into ALK targeted therapy. PLoS One. 2014;9(1):e84501. doi: 10.1371/journal.pone.0084501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Molecular cancer therapeutics. 2007;6(12 Pt 1):3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 58.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68(9):3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 59.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. The Lancet Oncology. 2012;13(10):1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013;368(25):2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 61.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England journal of medicine. 2014;371(23):2167–77. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 62.Toyokawa G, Seto T. Anaplastic lymphoma kinase rearrangement in lung cancer: its biological and clinical significance. Respiratory investigation. 2014;52(6):330–8. doi: 10.1016/j.resinv.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 63.Li S, Qi X, Huang Y, Liu D, Zhou F, Zhou C. Ceritinib (LDK378): A Potent Alternative to Crizotinib for ALK-Rearranged Non-Small-Cell Lung Cancer. Clinical lung cancer. 2015;16(2):86–91. doi: 10.1016/j.cllc.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 64.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. Journal of medicinal chemistry. 2013;56(14):5675–90. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 65.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer discovery. 2014;4(6):662–73. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014;370(13):1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ceritinib gains FDA approval for lung cancer. Cancer discovery. 2014;4(7):753–4. doi: 10.1158/2159-8290.CD-NB2014-074. [DOI] [PubMed] [Google Scholar]
- 68.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer cell. 2011;19(5):679–90. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 69.Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. The Lancet Oncology. 2014;15(10):1119–28. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- 70.Crystal AS, Shaw AT. Variants on a theme: a biomarker of crizotinib response in ALK-positive non-small cell lung cancer? Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(17):4479–81. doi: 10.1158/1078-0432.CCR-12-1952. [DOI] [PubMed] [Google Scholar]
- 71.Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(17):4682–90. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 72.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–6. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012;4(120):120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. The New England journal of medicine. 2010;363(18):1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 76.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011;71(18):6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(22):6219–26. doi: 10.1158/1078-0432.CCR-12-0392. [DOI] [PubMed] [Google Scholar]
- 78.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(5):1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rossing HH, Grauslund M, Urbanska EM, Melchior LC, Rask CK, Costa JC, et al. Concomitant occurrence of EGFR (epidermal growth factor receptor) and KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations in an ALK (anaplastic lymphoma kinase)-positive lung adenocarcinoma patient with acquired resistance to crizotinib: a case report. BMC research notes. 2013;6:489. doi: 10.1186/1756-0500-6-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20(9):1027–34. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(22):5686–96. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ou SH, Greenbowe J, Khan ZU, Azada MC, Ross JS, Stevens PJ, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung cancer. 2015 doi: 10.1016/j.lungcan.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 83.Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, et al. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J Thorac Oncol. 2014;9(12):1821–5. doi: 10.1097/JTO.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 84.Kim S, Kim TM, Kim DW, Go H, Keam B, Lee SH, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol. 2013;8(4):415–22. doi: 10.1097/JTO.0b013e318283dcc0. [DOI] [PubMed] [Google Scholar]
- 85.Cardarella S, Ortiz TM, Joshi VA, Butaney M, Jackman DM, Kwiatkowski DJ, et al. The introduction of systematic genomic testing for patients with non-small-cell lung cancer. J Thorac Oncol. 2012;7(12):1767–74. doi: 10.1097/JTO.0b013e3182745bcb. Epub 2012/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Graziano SL, Gamble GP, Newman NB, Abbott LZ, Rooney M, Mookherjee S, et al. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol. 1999;17(2):668–75. doi: 10.1200/JCO.1999.17.2.668. Epub 1999/03/18. [DOI] [PubMed] [Google Scholar]
- 87.Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26(26):4268–75. doi: 10.1200/JCO.2007.14.8924. Epub 2008/07/16. [DOI] [PubMed] [Google Scholar]
- 88.Brugger W, Triller N, Blasinska-Morawiec M, Curescu S, Sakalauskas R, Manikhas GM, et al. Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(31):4113–20. doi: 10.1200/JCO.2010.31.8162. Epub 2011/10/05. [DOI] [PubMed] [Google Scholar]
- 89.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. The Lancet Oncology. 2008;9(10):962–72. doi: 10.1016/S1470-2045(08)70206-7. Epub 2008/09/23. [DOI] [PubMed] [Google Scholar]
- 90.Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung cancer. 2010;69(3):272–8. doi: 10.1016/j.lungcan.2009.11.020. Epub 2009/12/22. [DOI] [PubMed] [Google Scholar]
- 91.Slebos RJ, Kibbelaar RE, Dalesio O, Kooistra A, Stam J, Meijer CJ, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. The New England journal of medicine. 1990;323(9):561–5. doi: 10.1056/NEJM199008303230902. [DOI] [PubMed] [Google Scholar]
- 92.Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, Siegfried JM. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clinical cancer research: an official journal of the American Association for Cancer Research. 1996;2(2):411–8. [PubMed] [Google Scholar]
- 93.Villaruz LC, Socinski MA, Cunningham DE, Chiosea SI, Burns TF, Siegfried JM, et al. The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer. 2013;119(12):2268–74. doi: 10.1002/cncr.28039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu HA, Sima CS, Shen R, Kass S, Gainor J, Shaw A, et al. Prognostic Impact of KRAS Mutation Subtypes in 677 Patients with Metastatic Lung Adenocarcinomas. J Thorac Oncol. 2015;10(3):431–7. doi: 10.1097/JTO.0000000000000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(15):4040–5. doi: 10.1158/1078-0432.CCR-12-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochimica et biophysica acta. 2009;1795(1):37–52. doi: 10.1016/j.bbcan.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 97.Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(16):4449–57. doi: 10.1158/1078-0432.CCR-11-3351. [DOI] [PubMed] [Google Scholar]
- 98.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014;371(21):1963–71. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(17):4570–9. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A. 2013;110(48):19519–24. doi: 10.1073/pnas.1319583110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 102.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70. doi: 10.1200/JCO.2011.35.6345. Epub 2012/01/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen YF, Hsieh MS, Wu SG, Chang YL, Shih JY, Liu YN, et al. Clinical and the prognostic characteristics of lung adenocarcinoma patients with ROS1 fusion in comparison with other driver mutations in East Asian populations. J Thorac Oncol. 2014;9(8):1171–9. doi: 10.1097/JTO.0000000000000232. [DOI] [PubMed] [Google Scholar]
- 104.Awad MM, Engelman JA, Shaw AT. Acquired resistance to crizotinib from a mutation in CD74-ROS1. The New England journal of medicine. 2013;369(12):1173. doi: 10.1056/NEJMc1309091. [DOI] [PubMed] [Google Scholar]
- 105.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31(16):1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 106.Klapper LN, Glathe S, Vaisman N, Hynes NE, Andrews GC, Sela M, et al. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A. 1999;96(9):4995–5000. doi: 10.1073/pnas.96.9.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 108.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 109.Arcila ME, Chaft JE, Nafa K, Roy-Chowdhuri S, Lau C, Zaidinski M, et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(18):4910–8. doi: 10.1158/1078-0432.CCR-12-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431(7008):525–6. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 111.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65(5):1642–6. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 112.Liu L, Shao X, Gao W, Bai J, Wang R, Huang P, et al. The role of human epidermal growth factor receptor 2 as a prognostic factor in lung cancer: a meta-analysis of published data. J Thorac Oncol. 2010;5(12):1922–32. doi: 10.1097/jto.0b013e3181f26266. [DOI] [PubMed] [Google Scholar]
- 113.Hirsch FR, Helfrich B, Franklin WA, Varella-Garcia M, Chan DC, Bunn PA., Jr Preclinical studies of gemcitabine and trastuzumab in breast and lung cancer cell lines. Clinical breast cancer. 2002;3(Suppl 1):12–6. doi: 10.3816/cbc.2002.s.003. [DOI] [PubMed] [Google Scholar]
- 114.Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, et al. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2004;15(1):19–27. doi: 10.1093/annonc/mdh031. [DOI] [PubMed] [Google Scholar]
- 115.Ross HJ, Blumenschein GR, Jr, Aisner J, Damjanov N, Dowlati A, Garst J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(6):1938–49. doi: 10.1158/1078-0432.CCR-08-3328. [DOI] [PubMed] [Google Scholar]
- 116.Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, et al. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene. 1996;12(8):1821–6. [PubMed] [Google Scholar]
- 117.Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30(35):4352–9. doi: 10.1200/JCO.2012.44.1477. [DOI] [PubMed] [Google Scholar]
- 118.Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome research. 2012;22(3):436–45. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–7. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yokota K, Sasaki H, Okuda K, Shimizu S, Shitara M, Hikosaka Y, et al. KIF5B/RET fusion gene in surgically-treated adenocarcinoma of the lung. Oncology reports. 2012;28(4):1187–92. doi: 10.3892/or.2012.1908. [DOI] [PubMed] [Google Scholar]
- 121.Pan Y, Zhang Y, Li Y, Hu H, Wang L, Li H, et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung cancer. 2014;84(2):121–6. doi: 10.1016/j.lungcan.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 122.Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suzuki M, Makinoshima H, Matsumoto S, Suzuki A, Mimaki S, Matsushima K, et al. Identification of a lung adenocarcinoma cell line with CCDC6-RET fusion gene and the effect of RET inhibitors in vitro and in vivo. Cancer science. 2013;104(7):896–903. doi: 10.1111/cas.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matsubara D, Kanai Y, Ishikawa S, Ohara S, Yoshimoto T, Sakatani T, et al. Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol. 2012;7(12):1872–6. doi: 10.1097/JTO.0b013e3182721ed1. [DOI] [PubMed] [Google Scholar]
- 125.Mukhopadhyay S, Pennell NA, Ali SM, Ross JS, Ma PC, Velcheti V. RET-rearranged lung adenocarcinomas with lymphangitic spread, psammoma bodies, and clinical responses to cabozantinib. J Thorac Oncol. 2014;9(11):1714–9. doi: 10.1097/JTO.0000000000000323. [DOI] [PubMed] [Google Scholar]
- 126.Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer discovery. 2013;3(6):630–5. doi: 10.1158/2159-8290.CD-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116(10):2695–706. doi: 10.1172/JCI28656. Epub 2006/08/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6(7):1152–61. doi: 10.1097/JTO.0b013e318216ee52. Epub 2011/05/21. [DOI] [PubMed] [Google Scholar]