

Abstract
The aim of this study was to uncover that unfolded protein response (UPR) contributed to the development of cisplatin resistance in osteosarcoma. MG-63 cells and SaOS-2 cells were exposed to cisplatin at presence or absence of 4-phenylbutyrayte (4-pba) and then analyzed by MTT assay and flow cytometry to determine the cell survival rates and apoptosis. Levels of glucose regulated protein 78KD (GRP78), C/EBP homologus protein (CHOP), cytoplasmic and nuclear NF-κB were detected by Western blot. Further, MG-63 cells and SaOS-2 cells were subjected to cisplatin with or without Bay 11-7082, a well-known inhibitor of NF-κB. After that, MTT assay and flow cytometry were used to determine the cell survival rates and apoptosis. Cisplatin and 4-PBA co-treatment significantly enhanced the cell apoptosis. Administration of cisplatin substantially increased the levels of GRP78 and CHOP. Moreover, mechanistic investigation uncovered that cisplatin promoted the levels of nuclear NF-κB whereas 4-PBA administration suppressed the cisplatin-induced accumulation of nuclear NF-κB level in osteosarcoma cells. Cisplatin combined with Bay 11-7082 obviously augmented MG-63 cells and SaOS-2 cells apoptosis when compared to that in osteosarcoma cells treated by cisplatin alone. Taken together, our data show that UPR protects osteosarcoma from cisplatin-mediated apoptosis through activation of NF-κB pathway. Therefore, targeting UPR may be a potential strategy to improve the osteosarcoma therapy.
Keywords: Osteosarcoma, UPR, cisplatin, chemoresistance, NF-κB
Introduction
Osteosarcoma (OS) is the most common type of primary malignant bone tumor in children and adolescents [1]. It remains the second cause responsible for tumor-related death in young adults. Although therapeutic strategies such as local tumor excision combined with neo-adjuvant chemotherapy have greatly raised the 5-year survival rate in patient with localized OS in last 3 decades, OS patients who have a poor response to chemotherapy usually suffer from a deadly outcome [2,3]. OS can develop into the acquired drug resistance phenotypes associated with chemotherapy, which is the main challenge to overcome. Therefore, it is of great importance to understand the mechanism underlying the OS drug resistance.
Endoplasmic reticulum (ER) is a critical organelle of eukaryotic cells that plays an essential role in correct folding of nascent secreted or membrane-bound proteins, lipid biosynthesis and Ca2+ homeostasis [4]. However, when the balance of protein folding is disrupted, accumulation of unfolded or misfolded protein in ER lumen triggers ER stress. Cells initiate the unfolded protein response (UPR), an adaptive process to transmit signal from ER to nucleus, restoring the ER homeostasis [5,6]. Activation of UPR leads to not only the general translation suppression but also an increase in capacity of ER protein folding. Recently, activation of UPR has been suggested to play a cytoprotective role in a range of human malignancies such as breast cancer, gastric cancer, hepatocellular carcinoma cells, pancreatic cancer, lung cancer and multiple myeloma [7-10]. However, the influence of UPR in osteosarcoma remains poorly understood.
The nuclear factor κB (NF-κB) has been shown to be a key regulator in immune response, inflammation as well as survival, proliferation, metastasis and chemoresistance in cancer [11-13]. NF-κB is a dimer of the Rel family including RelA (p65), p50, p52, RelB and c-Rel. Each of them binds to their cognate response element as homo-or hetero-dimers with p65/p50 being the canonical heterodimers. Recently, activation of NF-κB leads to increase cell proliferation in osteosarcoma and hinder osteoblastic differentiation. In addition, blockage of NF-κB activation sensitizes osteosarcoma cell lines to chemotherapeutic agents [14,15].
Accordingly, we raise the hypothesis that initiation of UPR may contribute to the drug resistance of osteosarcoma to cisplatin by activation of NF-κB signal. In order to testify our hypothesis, we investigated the roles and mechanisms of UPR in chemoresistance of osteosarcoma to cisplatin. Attenuation of UPR activation sensitized osteosarcoma to cisplatin. Furthermore, we also unveiled that administration of cisplatin activated NF-κB and 4-PBA suppressed cisplatin-induced NF-κB activation. Blockage of NF-κB significantly promotes cisplatin-induced apoptosis in human osteosarcoma cells.
Materials and methods
Cell culture
Human osteosarcoma cell line MG-63 and SaOS-2 cells were obtained from the American Type Culture Collection (ATCC). The osteosarcoam cell lines were cultured in DMEM medium supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml), containing 10% (v/v) fetal bovine serum (FBS; Gibco BRL, Life Technologies, Carlsbad, CA). Cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Antibodies and chemicals
Thapsigargin (TG), dimethly sulfoxiase (DMSO), 4-phenylbutyrayte (4-PBA), Bay 11-7082 and cisplatin were obtained from Sigma (Sigma, St. Louis, MO). TG, Bay 11-7082 and 4-phenylbutyrayte were dissolved in DMSO. Cisplatin was dissolved in PBS. The antibody to GRP78 was purchased from Santa Cruz (Santa Cruz, CA). The antibodies to β-actin, PCNA, CHOP and NF-κB p65 were obtained from Cell signaling (Cell signaling, Beverly, MA). The horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Sigma (Sigma, St. Louis, MO).
Cell viability assays
The MTT assay was used to test cell viability. We seeded growing MG-63 cells into 96-well culture plates in 200 μl of media at a density of 1×104 cells/well. The following day, 200 μl media with indicated chemicals were added to each well and incubated for indicated hours. Following the incubation, 20 μl MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphe-nyltetrazolium bromide (5 mg/ml in PBS; Sigma, St. Louis, MO) was added to each well for 4 h. Subsequently, the supernatants were discarded and 150 μl DMSO was added to each well. After plates were shaken for 10 min in dark, absorbance was measured at a wavelength of 490 nm using a microplate reader (Multiskan GO, Thermo scientific, Waltham, MA). MG-63 and SaOS-2 cells were treated with indicated cisplatin concentrations for 24 hours. Then cells viability and IC50 of cisplatin were calculated. As 4-PBA has been used in various cells with different concentrations from 1 to 20 mmol/L, we first assessed the MG-63 viability by MTT assay after incubated with indicated concentration of 4-PBA for 24 hours. Then, we preferred to select 2.5 mmol/L, 5 mmol/L and 10 mmol/L 4-PBA to co-treat MG-63 cells with cisplatin.
Extraction of nuclear fractions
Nuclear protein extracts were prepared according to the previous protocol. Briefly, cells (1×106) were harvested after trypsinization, washed twice with cold PBS, and then lysed with cold buffer A (10 mmol/L HEPES, pH 7.9, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L ethylene glycol tetraacetic acid, 1 mmol/L phenyl methane sulfonyl fluoride (PMSF), 1 mmol/L dithiothreitol, 1 mg/L aprotinin, 1 mg/L leupeptin, and 1 mg/L pepstatin A). After incubation for 10 min on ice, 0.1% NP 40 was added to the lysates and the tubes were shaken for 10 seconds on vortex. The cell homogenates were centrifuged at 12,000 g for 5 min at 4°C. The supernatants (cytoplasmic extracts) were harvested and stored at -70°C. Next, the nuclear pellets were resuspended in cold buffer B (20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 0.1 mmol/L EDTA, 0.1 mmol/L ethylene glycol tetraacetic acid, 1 mmol/L phenylmethanesulfonylfluoride, 1 mmol/L dithiothreitol, 1 mg/ L aprotinin, 1 mg/ L leupeptin, and 1 mg/L pepstatin A) and shaken at maximum speed at 4°C for 30 min on vortex. The lystates were centrifuged at 12,000 g for 5 min, and the supernatants (nuclear extract) were stored at -70°C. For western blotting analysis, total protein concentration was measured by the BCA assay (Pierce, Rockford, IL, USA).
Western blot analysis
Cell lysates were prepared with cell lysis buffer (RIPA buffer, 10 mmol/L Tris-HCl, pH 8.0, 10 mmol/L EDTA, 0.15 mol/L NaCl, 1% NP-40, 0.5% sodium dodecyl sulphate, 1 μg/ml aprotinin, 1 mmol/L PMSF) on ice for 30 min. The protein supernatants were collected by centrifugation at 10,000 g for 10 min. Total protein concentration was measured by the BCA assay. Each extract containing approximately 25-30 μg protein was subjected to the 10% polyacrylmide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) by wet transfer. The membranes were blocked by TBS-T (10 mmol/L Tris-HCl, pH 7.4, 100 mmol/L NaCl, 0.1% Tween 20) containing 5% nonfat dry milk for 2 h at room temperature and incubated with primary antibodies (GRP78, 1:800 dilution; CHOP, 1:1000 dilution; β-actin,1:1000 dilution; NF-κB p65,1:1000 dilution; PCNA, 1:1000 dilution) overnight at 4°C. After three washes by TBS-T for 15 min, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies. The blotting signals were detected by enhanced chemiluminescence (Pierce, Rockford, IL). β-actin and PCNA were acted as loading control.
Apoptosis detection by flow cytometry
The apoptotic cells were measured by Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Diego, USA). Briefly, 1×105 cells were seeded in 6-well plates and then incubated with indicated chemicals for indicated time. After treatment, approximately 1×106 cells were collected, washed twice with cold PBS, and then stained with Annexin V-FITC and PI according to the manufacturer’s instructions. The resulting fluorescence was analyzed by flow cytometry (BD FACS Jazz, San Jose, CA).
Statistical analysis
Statistical analyses were performed using the SPSS 17.0 Statistical software. All data were presented as the mean ± SD Statistical comparisons were performed using one-way ANOVA. When significant values were found (P<0.05), the Turkey’s Honestly Significant Differences tests were used to compare the differences in two groups.
Results
Attenuation of ER stress promotes cisplatin-induced MG-63 cells and SaOS-2 cells apoptosis
As the UPR is of great importance to protect cells from apoptosis under stress, we explored the potential role of cisplatin-induced UPR in osteosarcoma cells. As showed in Figure 1A, the IC50 of cisplatin at MG-63 and SaOS-2 were 46.5 μmol/L and 39.1 μmol/L respectively. We prefer cisplatin at the concentration of 20 μmol/L in following study, because our previous report used cisplatin at 20 μmol/L [16]. Figure 1B and 1E indicated that 4-PBA had an obviously cytotoxic effect on MG-63 cells and SaOS-2 cells at 20 mmol/L, whereas 4-PBA at the concentration of 2.5 mmol/L, 5 mmol/L and 10 mmol/L did not cause significant cell death. Then, 4-PBA treatment with the concentration of 2.5 mmol/L, 5 mmol/L and 10 mmol/L led to MG-63 cells and SaOS-2 cells more sensitive to cisplatin induced cell injury (Figure 1C and 1F) and promoted cell apoptotic cell death (Figure 1D and 1G). These data show that attenuation of ER stress promotes cisplatin-induced MG-63 cells and SaOS-2 cells apoptosis.
Figure 1.

Attenuation of ER stress promoted cisplatin induced osteosarcoma cells apoptosis. A: Indicated osteosarcoma cells were treated with different concentration of cisplatin for 24 hours, then the cell viability was analyzed by MTT (n = 4). B and E: MG-63 cells and SaOS-2 cells were treated with indicated concentrations of 4-PBA for 24 hours and then cell viability was analyzed by MTT assay respectively (n = 3). C and F: MG-63 cells and SaOS-2 cells were treated with cisplatin (20 μmol/L) in the presence or absence of indicated concentrations of 4-PBA for 24 hours. Cell viability was analyzed by MTT assay (n = 3). D and G: MG-63 cells and SaOS-2 cells were treated with cisplatin (20 μmol/L) in the presence or absence of 4-PBA (10 mmol/L) for 24 respectively. Apoptosis was analyzed by the Annexin V assay by flow cytometry (n = 3). Data are represented as mean ± SD. *P<0.05, **P<0.01 compared to control; &*P<0.05, &&*P<0.01 compared to cisplatin alone.
Cisplatin induces UPR in MG-63 and SaOS-2 cells
To explore whether cisplatin is able to trigger UPR induction in osteosarcoma cells, MG-63 cells and SaOS-2 cells were treated with cisplatin for indicated time. TG administration was used as positive control. Cisplatin treatment substantially elevated the expression of GRP78 (Figure 2A and 2C) and CHOP (Figure 2B and 2D) in MG-63 and SaOS-2 cells respectively. Then, MG-63 and SaOS-2 cells were treated with cisplatin in the presence or absence of 4-PBA (10 mmol/L). As expected, adminstration of 4-PBA reversed the increased GRP78 (Figure 3A and 3C) and CHOP expression (Figure 3B and 3D) in MG-63 and SaOS-2 cells respectively. Therefore, these findings show that cisplatin induces UPR activation in osteosarcoma cells.
Figure 2.

Effect of Cisplatin on levels of GRP78 and CHOP protein in osteosarcoma cells. MG-63 cells and SaOS-2 cells were treated with cisplatin (20 μmol/L) for indicated periods and TG (0.01 μmol/L) for positive control. Representative Western blot analysis of GRP78 protein level (A) and CHOP protein level (B) in OS cells treated with cisplatin. (C and D) Analyses of band intensity are shown as the relative ratio of GRP (C) and CHOP (D) to actin Data are represented as mean ± SD. Β-actin was served as load control. *P<0.05, #P<0.01 versus control. All figures are representatives of the results of three independent experiments.
Figure 3.

Effects of 4-PBA on the levels of GRP78 and CHOP protein in cisplatin-treated OS cells. MG-63 and SaOS-2 cells were treated with cisplatin (20 μmol/L) for 24 hours in the presence or absence 4-PBA (10 mmol/L). Representative Western blot analysis of GRP78 protein level (A) and CHOP protein level (B) in OS cells. Analyses of band intensity are shown as the relative ratio of GRP (C) and CHOP (D) to actin. Data are represented as mean ± SD. β-actin was served as load control. #P<0.05 versus control; *P<0.05 versus cisplatin. All figures are representatives of the results of three independent experiments.
NF-κB is activated in OS cells treated with cisplatin
To validate whether NF-κB signaling is activated in MG-63 cells and SaOS-2 cells treated with cisplatin, the levels of NF-κB p65 in cytoplasm and nucleus were monitored. Administration of cisplatin induced a moderate decrease of NF-κB p65 subunit level in cytoplasm (Figure 4A) and accumulation of NF-κB p65 subunit inside the nucleus (Figure 4B). In order to validate that NF-κB activation was related with UPR induction, we evaluated the activation of NF-κB in MG-63 and SaOS-2 cells following 4-PBA treatment. As expected, 4-PBA treatment alone displayed little effect on the levels of cytoplasmic and nuclear p65 protein in OS cells. Figure 4C and 4D showed that adminstration of 4-PBA decreased the levels of nuclear p65 protein and increased the levels of cytoplasmic p65 protein in cisplatin-treated OS cells. These finding supports the fact that NF-κB singal is activated in cisplatin-treated MG-63 and SaOS-2 cells.
Figure 4.
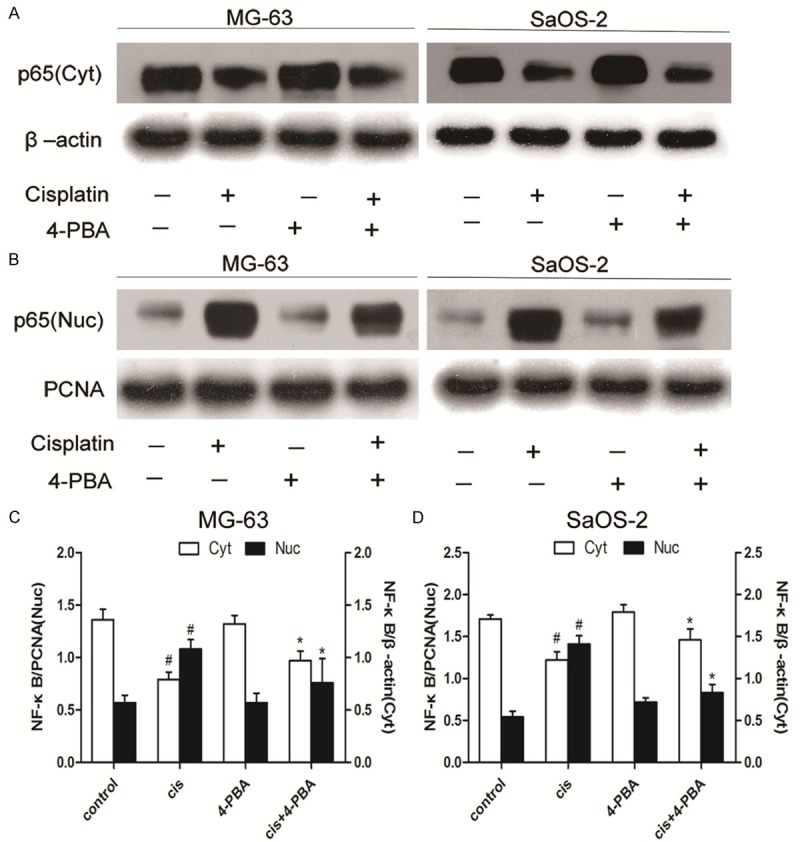
Effects of 4-PBA on the activations of NF-κB p65 in cisplatin-treated osteosarcoma cells. MG-63 and SaOS-2 cells were treated with cisplatin (20 μmol/L) for 24 hours in the presence or absence of 4-PBA (10 mmol/L). Representatives Western blots of cytoplasmic NF-κB p65 subunit level (A) and nuclear NF-κB p65 subunit level (B) in OS cells. Analyses of band intensity are shown as the relative ratio of cytoplasmic NF-κB p65 to β-actin and nuclear NF-κB p65 to PCNA in MG-63 cells (C). Analyses of band intensity are shown as the relative ratio of cytoplasmic NF-κB p65 to β-actin and nuclear NF-κB p65 to PCNA in SaOS-2 cells (D). PCNA was served as nuclear load control. β-actin was served as cytoplasmic load control. Data are represented as mean ± SD. Figures of NF-κB p65 subunit are representatives of the results from three independent experiments. #P<0.05 versus control; *P<0.05 versus cisplatin.
NF-κB activation contributes to the cytoprotective role of UPR in cisplatin-treated OS cells
To validate whether NF-κB signaling contributes to the cytoprotective role of UPR in cisplatin-treated osteosarcoma cells, MG-63 and SaOS-2 cells were pretreated with Bay 11-7082, a well-known inhibitor of NF-κB. No significant cell death was observed after MG-63 and SaOS-2 cells incubated with indicated concentration of Bay 11-7082 alone (Figure 5A and 5D). Figure 5B indicated that co-treatment with cisplatin and Bay 11-7082 at 5, 10 μmol/L showed no significant difference in cell viability when compared with that in cisplatin treatment alone in MG-63 cells. Indeed, Bay 11-7082 treatment at the concentration 20 μmol/L led to MG-63 cells more sensitive to cisplatin induced cell injury. Further, pretreatment with Bay 11-7082 (20 μmol/L) obviously promoted the cisplatin-induced MG-63 cell apoptosis (Figure 5C). Figure 5E indicated SaOS-2 cells were more sensitive to cisplatin induced cell injury after Bay 11-7082 addition. Cisplatin-induced Sa-OS2 cells apoptosis was markedly increased following Bay 11-7082 administration (Figure 5F). These data suggest NF-κB activation protects MG63 and SaOS-2 cells from cisplatin induced apoptosis.
Figure 5.
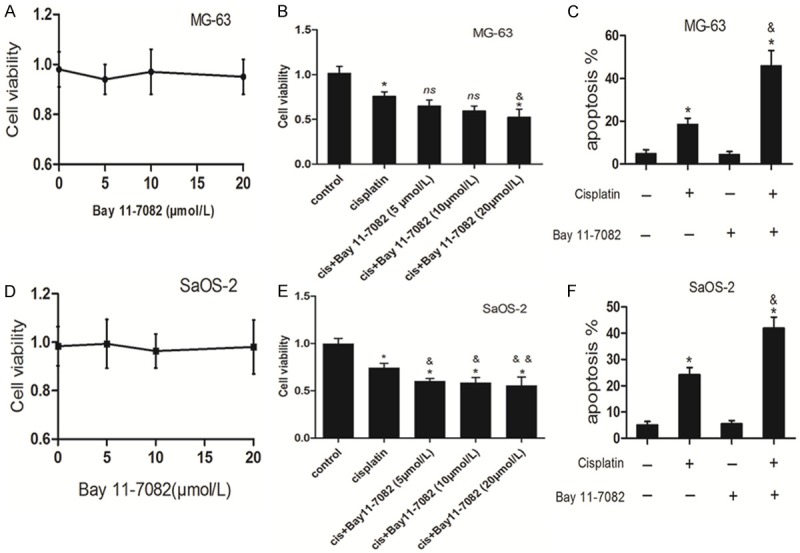
Activation of NF-κB contributes to the cisplatin-treated osteosarcoma cells. MG-63 cells (A) and SaOS-2 cells (D) were treated with indicated concentrations of Bay 11-7082 alone for 24 hours and then cell viability was analyzed by MTT assay respectively (n = 3). MG-63 cells (B) and SaOS-2 cells (E) were treated with cisplatin (20 μmol/L) in the presence or absence of indicated concentrations of Bay 11-7082 for 24 hours. Cell viability was analyzed by MTT assay (n = 3). MG-63 cells (C) and SaOS-2 cells (F) were treated with cisplatin (20 μmol/L) in the presence or absence of Bay 11-7082 (20 μmol/L) for 24 respectively. Apoptosis was analyzed by the Annexin V assay by flow cytometry (n = 3). Data are represented as mean ± SD. *P<0.05, **P<0.01 compared to control; &*P<0.05, &&*P<0.01 compared to cisplatin alone; ns = no significance.
Discussion
Although the therapeutic approaches such as neoadjuvant chemotherapy and surgical resection have developed rapidly, the cure rate of osteosarcoma still remains low. The developed drug resistance of OS is becoming a tough barrier to achieve the satisfied outcomes [17]. Drug resistance of OS attributes to various mechanisms including reduced intracellular drug concentration, dysfunction of membrane transport, inactivation of drug, anti-apoptosis, increased DNA repair and stem cell-like cancer cells induced chemoresistance [18]. In this study, we presented that UPR activation was another contributor to cisplatin resistance in OS. Mitigation of UPR sensitized OS cells to cisplatin. Meanwhile, activation of NF-κB pathway contributed to the cytoprotective role of UPR in OS cells.
Cisplatin, a first class anti-tumor drug widely used in various cancers, targets the DNA to induce apoptosis through mitochondrial death pathway or Fas death pathway [19,20]. Cisplatin-induced ER stress had been reported in human hepatocellular carcinoma cells and melanoma cells [21,22]. Our study shows that cisplatin treatment induces the UPR activation. Moreover, the findings that 4-PBA mitigated cisplatin-induced increased expression of GRP78 and CHOP, provide additional support for this phenomenon. This observation consisted with previous report that indicated cisplatin treatment induced ER stress in rat kidney tissue in vivo [23]. It has been clear that activation of calpain in cisplatin manner is responsible for cisplatin induced ER stress [22].
In our study, attenuation of ER stress promoted cisplatin induced OS cells apoptosis. These results are in accord with recent reports [21-24]. It has been validated that UPR favors cancer cells adaption, escaping from cell demise and persistent growth in massive types of malignancies [25-27]. UPR aims to restore ER homeostasis during ER stress. However, if ER stress is severe and prolonger, cancer cells will succumb to UPR mediated apoptosis [28,29]. The fact that UPR is cytoprotective as well as being cytotoxic has been supported by numerous evidences. For instance, the spliced X-boxing binding protein1 (XBP1s), downstream of IRE1, has been shown to contribute to the angiogenesis, proliferation and drug resistance in tumors [30]. ATF6 activation enhances the transcription of genes encoding chaperones and enzymes that assist protein folding, which alleviates ER stress and improves the resistance of tumor to ER stress induced death [31,32]. Meanwhile, ATF4, downstream of PERK/eukaryotic initiation factor 2α (eIF2α), promotes hypoxia resistance in tumors [33]. However, PERK dependent activation of CHOP dominates and leads to cell apoptosis in severe and prolonger ER stress [34]. Therefore, the double-edged sword role of UPR in cancer cells fate determination may depend on cell type and ER stress severity. The strategies that target UPR has mainly divided into two types: (1) enhancing misfolded proteins in ER lumen to initiate much more severe ER stress; (2) blocking UPR dependent pro-survival effect. Fortunately, these approaches have been confirmed to be effective to override the drug resistance in tumors [35]. In our study, UPR activation acts as a mechanism of survival in OS and targeting UPR shows a promising approach to overcome drug resistance in OS.
As NF-κB plays a critical role in pro-survival and drug resistance in osteosarcoma cells [15], it is logical to present the hypothesis that NF-κB signal is activated and protects osteosarcoma cells from cisplatin cytotoxicity. Our data suggest adminstration of cisplatin activates NF-κB and this activation is suppressed following 4-PBA treatment. This phenomenon indicates that UPR contributes to NF-κB activation, which consists with previous reports [36-38]. It has been clear that the PERK and IRE1, two arms of UPR, are responsible for NF-κB activation. When UPR is initiated, phosphorylated eIF2α by PERK suppresses general translation including the IκB and NF-κB. Because IκB is characterized with shorter half-live than that in NF-κB, general translational suppression augments the ratio of NF-κB to IκB. Therefore, NF-κB dissociates from IκB blockage and translocates into nucleus [36]. In response to ER stress, IRE1 undergoes autophoshorylation and binds to the adaptor tumor-necrosis factor-α-receptor-associated factor 2 (TRAF2). It has been found that IRE1/TRAF2 complex recruits and phosohorylates IKK. After that, IKK phosphorylates IκB and elicits the ubquitination and degradation of IκB, leading to the nuclear delivery of NF-κB [37]. Recently, the interplay of PERK and IER1 activities was found to be essential for activation of NF-κB in response to ER stress. Indeed, without IRE1 mediated basal phosphorylation of IKK, phosphorylated eIF2α induced general translational suppression fails to decrease IκB level enough to set a substantial number of NF-κB free [39]. In our study, incubation with Bay 11-7082, an inhibitor of IKK, promotes cisplatin-induced OS cells apoptosis, which consisted with previous report [15]. It has been unveiled that NF-κB contributes to anti-apoptosis and tumorigenesis through regulation of Bcl-xl, cFLIP and cIAP 1, cIAP2, cyclin D1, c-myc and Bfl-1 [40-42]. Some drugs that targeting NF-κB signal has been demonstrated to sensitize tumors to anti-tumor agents [43-45].
In conclusion, treatment with cisplatin is able to induce UPR in OS cells and targeting UPR sensitizes OS cells to cisplatin. The cytoprotective role of UPR in OS cells may be through activation of NF-κB pathway. Thus, these data suggest that targeting UPR and NF-κB may be a potential strategy for osteosarcoma therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 81272947 and 81302338). Dr. Jun Wang contributes to the revision of the English.
Disclosure of conflict of interest
None.
References
- 1.Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res. 2010;5:78. doi: 10.1186/1749-799X-5-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47:2431–2445. doi: 10.1016/j.ejca.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Zhang W. New molecular insights into osteosarcoma targeted therapy. Curr Opin Oncol. 2013;25:398–406. doi: 10.1097/CCO.0b013e3283622c1b. [DOI] [PubMed] [Google Scholar]
- 4.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 6.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 7.Al-Rawashdeh FY, Scriven P, Cameron IC, Vergani PV, Wyld L. Unfolded protein response activation contributes to chemoresistance in hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2010;22:1099–1105. doi: 10.1097/MEG.0b013e3283378405. [DOI] [PubMed] [Google Scholar]
- 8.Scriven P, Coulson S, Haines R, Balasubramanian S, Cross S, Wyld L. Activation and clinical significance of the unfolded protein response in breast cancer. Br J Cancer. 2009;101:1692–1698. doi: 10.1038/sj.bjc.6605365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uramoto H, Sugio K, Oyama T, Nakata S, Ono K, Yoshimastu T, Morita M, Yasumoto K. Expression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significance. Lung Cancer. 2005;49:55–62. doi: 10.1016/j.lungcan.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L, Wei D, Yao J, Fang S, Xie K. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastasis. 2006;23:401–410. doi: 10.1007/s10585-006-9051-9. [DOI] [PubMed] [Google Scholar]
- 11.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 13.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 14.Andela VB, Sheu TJ, Puzas EJ, Schwarz EM, O’Keefe RJ, Rosier RN. Malignant reversion of a human osteosarcoma cell line, Saos-2, by inhibition of NFkappaB. Biochem Biophys Res Commun. 2002;297:237–241. doi: 10.1016/s0006-291x(02)02141-1. [DOI] [PubMed] [Google Scholar]
- 15.Castro-Gamero AM, Borges KS, Da Silva Silveira V, Lira RCP, de Paula Gomes Queiroz R, Valera FCP, Scrideli CA, Umezawa K, Tone LG. Inhibition of nuclear factor-κB by dehydroxymethylepoxyquinomicin induces schedule-dependent chemosensitivity to anticancer drugs and enhances chemoinduced apoptosis in osteosarcoma cells. Anti-Cancer Drugs. 2012;23:638–650. doi: 10.1097/CAD.0b013e328350e835. [DOI] [PubMed] [Google Scholar]
- 16.Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72:230–238. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 17.Jaffe N. Historical perspective on the introduction and use of chemotherapy for the treatment of osteosarcoma. Adv Exp Med Biol. 2014;804:1–30. doi: 10.1007/978-3-319-04843-7_1. [DOI] [PubMed] [Google Scholar]
- 18.He H, Ni J, Huang J. Molecular mechanisms of chemoresistance in osteosarcoma (Review) Oncol Lett. 2014;7:1352–1362. doi: 10.3892/ol.2014.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinato O, Musetti C, Sissi C. Pt-based drugs: the spotlight will be on proteins. Metallomics. 2014;6:380–395. doi: 10.1039/c3mt00357d. [DOI] [PubMed] [Google Scholar]
- 20.Rebillard A, Lagadic-Gossmann D, Dimanche-Boitrel MT. Cisplatin cytotoxicity: DNA and plasma membrane targets. Curr Med Chem. 2008;15:2656–2663. doi: 10.2174/092986708786242903. [DOI] [PubMed] [Google Scholar]
- 21.Chen R, Dai RY, Duan CY, Liu YP, Chen SK, Yan DM, Chen CN, Wei M, Li H. Unfolded protein response suppresses cisplatin-induced apoptosis via autophagy regulation in human hepatocellular carcinoma cells. Folia Biol (Praha) 2011;57:87–95. doi: 10.14712/fb2011057030087. [DOI] [PubMed] [Google Scholar]
- 22.Mandic A. Cisplatin Induces Endoplasmic Reticulum Stress and Nucleus-independent Apoptotic Signaling. J Biol Chem. 2003;278:9100–9106. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 23.Peyrou M, Hanna PE, Cribb AE. Cisplatin, Gentamicin, and p-Aminophenol Induce Markers of Endoplasmic Reticulum Stress in the Rat Kidneys. Toxicol Sci. 2007;99:346–353. doi: 10.1093/toxsci/kfm152. [DOI] [PubMed] [Google Scholar]
- 24.Feng R, Zhai WL, Yang HY, Jin H, Zhang QX. Induction of ER stress protects gastric cancer cells against apoptosis induced by cisplatin and doxorubicin through activation of p38 MAPK. Biochem Biophys Res Commun. 2011;406:299–304. doi: 10.1016/j.bbrc.2011.02.036. [DOI] [PubMed] [Google Scholar]
- 25.Matsumura K, Sakai C, Kawakami S, Yamashita F, Hashida M. Inhibition of cancer cell growth by GRP78 siRNA lipoplex via activation of unfolded protein response. Biol Pharm Bull. 2014;37:648–53. doi: 10.1248/bpb.b13-00930. [DOI] [PubMed] [Google Scholar]
- 26.Yadav RK, Chae S, Kim H, Chae HJ. Endoplasmic Reticulum Stress and Cancer. J Cancer Prev. 2014;19:75–88. doi: 10.15430/JCP.2014.19.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2012;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandewynckel YP, Laukens D, Geerts A, Bogaerts E, Paridaens A, Verhelst X, Janssens S, Heindryckx F, Van Vlierberghe H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013;33:4683–4694. [PubMed] [Google Scholar]
- 30.Shajahan AN, Riggins RB, Clarke R. The role of X-box binding protein-1 in tumorigenicity. Drug News Perspect. 2009;22:241. doi: 10.1358/dnp.2009.22.5.1378631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 32.Belmont PJ, Chen WJ, Thuerauf DJ, Glembotski CC. Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J Mol Cell Cardiol. 2012;52:1176–1182. doi: 10.1016/j.yjmcc.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5:723–728. doi: 10.4161/cbt.5.7.2967. [DOI] [PubMed] [Google Scholar]
- 34.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta. 2013;1833:1356–1366. doi: 10.1016/j.bbamcr.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prell T, Lautenschläger J, Weidemann L, Ruhmer J, Witte OW, Grosskreutz J. Endoplasmic reticulum stress is accompanied by activation of NF-κB in amyotrophic lateral sclerosis. J Neuroimmunol. 2014;270:29–36. doi: 10.1016/j.jneuroim.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Tam AB, Mercado EL, Hoffmann A, Niwa M. ER Stress Activates NF-κB by Integrating Functions of Basal IKK Activity, IRE1 and PERK. PLoS One. 2012;7:e45078. doi: 10.1371/journal.pone.0045078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Q, Lee HH, Li Y, Parks TP, Cheng G. Upregulation of Bcl-x and Bfl-1 as a potential mechanism of chemoresistance, which can be overcome by NF-kappaB inhibition. Oncogene. 2000;19:4936–4940. doi: 10.1038/sj.onc.1203861. [DOI] [PubMed] [Google Scholar]
- 41.Cosimo E, McCaig AM, Carter-Brzezinski LJM, Wheadon H, Leach MT, Le Ster K, Berthou C, Durieu E, Oumata N, Galons H, Meijer L, Michie AM. Inhibition of NF-B-Mediated signaling by the cyclin-dependent Kinase inhibitor CR8 overcomes prosurvival stimuli to induce apoptosis in chronic lymphocytic leukemia cells. Clin Cancer Res. 2013;19:2393–2405. doi: 10.1158/1078-0432.CCR-12-2170. [DOI] [PubMed] [Google Scholar]
- 42.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death-a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin SH, Kim TI, Han DS, Shin SK, Kim WH. Thalidomide suppresses the interleukin 1beta-induced NFkappaB signaling pathway in colon cancer cells. Ann N Y Acad Sci. 2002;973:414–418. doi: 10.1111/j.1749-6632.2002.tb04674.x. [DOI] [PubMed] [Google Scholar]
- 44.Yu J, Qian H, Li Y, Wang Y, Zhang X, Liang X, Fu M, Lin C. Arsenic trioxide (As2O3) reduces the invasive and metastatic properties of cervical cancer cells in vitro and in vivo. Gynecol Oncol. 2007;106:400–406. doi: 10.1016/j.ygyno.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 45.Almawi WY, Melemedjian OK. Negative regulation of nuclear factor-kappaB activation and function by glucocorticoids. J Mol Endocrinol. 2002;28:69–78. doi: 10.1677/jme.0.0280069. [DOI] [PubMed] [Google Scholar]