

Abstract
Acid sensing ion channels (ASICs), activated by lowering extracellular pH, play an important role in normal synaptic transmission in brain and in the pathology of brain ischemia. ASICs activation involving in glutamate receptor-independent ischemic brain injury has been generally accepted, and PICK1 is recently shown to be one of partner proteins interacting with ASICs through its PDZ domain. Here we showed that ASICs and PICK1 played key roles in OGD-Rep process. In wild-type cultured cortical neurons, not only the amplitude of ASICs current and the calcium transients induced by acidosis were both increased after OGD-Rep, but also the total protein levels of ASIC1 and ASIC2a were up-regulated progressively after ischemia insults, indicating that ASICs play a vital role in neuronal ischemia. However, these activities were reversed with PICK1-knockout after OGD-Rep, accompanied with the dramatically down-regulating the protein abundances of ASIC1 and ASIC2a, which suggested the neuroprotection activity in brain ischemia by PICK1-knockout. These results indicate that knocking-out PICK1 gene casts the neuroprotection effect by reducing ASICs current and the calcium transients in OGD-Rep neuronal cells, which will offer a promising strategy in the therapy of brain ischemia.
Keywords: ASICs, PICK1, OGD-Rep, calcium transients, brain ischemia, neuroprotection
Introduction
Neuronal injury, which is induced by cerebral ischemia-reperfusion, is a very complex process associated with excitotoxicity, oxidative stress, apoptosis and necrosis [1]. Intracellular Ca2+ overload is an important factor of brain ischemia. Excessive Ca2+ in the cell activates a cascade of cytotoxic events leading to activation of enzymes that break down proteins, lipids, and nucleic acids [2,3]. Many in vivo studies indicate that acidosis aggravates ischemic brain injury [4]. Recent evidence suggests that activation of Ca2+-permeable amiloride-sensitive ASIC1a may be responsible for acidosis-mediated, glutamate receptor independent, neuronal injury [5,6]. These suggested a possible role for ASICs in mediating a cellular response to ischemia.
ASIC channels are expressed in large numbers by neurons of the central nervous system. To date, six members of the ASIC family are cloned and at least three (ASIC1a, ASIC2a, and ASIC2b) subunits were found in neurons [7,8]. Overactivation of ASICs contributes to neurodegenerative diseases such as ischemic brain/spinal cord injury, multiple sclerosis, Parkinson’s disease, and Huntington’s disease [9]. It is well known that ischemia results in a marked reduction in tissue pH, and that acidosis is an important determinant of neurological injury [10]. ASICs are activated as pH falls, and generally inactivated at physiological pH (pH 7.4) [9]. Homomeric ASIC1a and heteromeric ASIC1a/2b channels are able to conduct both Na+ and Ca2+ [4,11,12]. As members of the epithelial Na+ channel/degener in family of ion channels, ASICs have two transmembrane domains separated by a large extracellular loop, with cytoplasmic amino and C termini [7]. They can form homo- and heteromeric channels, different subunit combinations produce channels with various pH sensitivity, kinetics and permeation properties [13]. Prolonged exposure to an acidic environment may also alter the number of ASIC channels in central neurons [14], perhaps altering their responsiveness to acidosis.
PICK1 (protein interacting with C kinase 1) is a scaffolding protein that regulates trafficking of multiple membrane proteins [15]. Its PDZ domain mediates the interaction with both ASIC1a- and ASIC2a-intracellular C terminus via a protein kinase A-dependent process [16]. It has also been demonstrated that PICK-1 actually serves as an intermediary protein through which protein kinase C acts to up-regulate ASIC2a expression [17] and to modulate heteromultimeric ASIC2b-ASIC3 channel expression and pH sensitivity [18]. In a recent study, it showed that PICK1 increases ASIC1a-mediated acidotoxicity and this effect requires both the PDZ and BAR domains of PICK1 [19]. Disruption of PICK1 attenuates the function of ASICs [20].
There is no direct evidence to prove the effects of ASICs and PICK1 with ischemia-reperfusion injury so far, especially at molecular and cellular level. Therefore, we used the oxygen and glucose deprivation followed by reperfusion (OGD-Rep) model to investigate the effects of ASICs and PICK1 on primary cultured cortical neural cell injury. We demonstrate that activation of ASICs is largely responsible for ischemic neuronal injury and disrupting PICK1 gene exerts significant neuroprotective effect by downregulating ASICs current and the calcium transients during process of ischemia-reperfusion, which provides us a new gene target in the therapy of brain ischemia.
Materials and methods
Animals
C57/BL6 mice of PICK1+/+, +/- and -/- were kindly provided by Dr. Jun Xia and were mated to intercross. All animals were bred at 24°C in specific pathogen-free animal house and provided with standard mice chow and water ad libitum. The investigation conformed to the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH) of the United States and was approved by the Ethics Committee for the Use of Human or Animal Subjects of Huazhong University of Science and Technology.
Primary cortical neuron culture
Cultures of cortical neurons were prepared from postnatal day 0-1 littermate mice as previously [21] described with some modifications. Briefly, cortices of newborn mice were decapitated and brains were rapidly removed and placed into ice-cold PBS, and the tails were kept at -20°C in order to identify the genotype. Cerebral cortices were dissected, minced and digested with 0.125% trypsin in PBS for 18-20 min at 37°C in separate containers. Followed by repeated pipetting, and filtration through a falcon cell strainer and centrifugation at 118× g for 5 min, the cell pellets were re-suspended and plated on 0.01% poly-L-lysine coated 6-mm glass coverslips or 35-mm Petri dishes, 24- or 96- well plates. Isolated neurons were cultured with Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (Gibco, NY, USA), 2 mM L-glutamine (Sigma, MO, USA), and 100 U/ml penicillin-Streptomycin. After 1 day, the medium was changed to DMEM supplemented with 2% B27 (Gibco, NY, USA).
Genotyping of PICK1 knockout mice
Tails kept during cell cultures were collected to carry out the genotype identifications by RT-PCR analysis. The genome DNA was extracted from the tails and PCR was employed to amplify the PICK1 gene fragments. Five microliters of DNA were used as a template in each 25 µl PCR reactions. Amplifications were performed as follows: 40 cycles, at 95°C for 60 s, 60°C for 60 s, and 72°C for 40 s. PCR products were electrophoresed on a 2% agarose gel using PCR markers (Tiangen Biotech, China) as the standard to determine the molecular size. Analysis was performed GENIUS bio imaging system.
Neuronal ischemic-reperfusion model
The neuronal cells were grown as monolayers in tissue culture flasks under normal conditions. To initiate OGD, the cell culture medium was removed and the cells were washed twice with glucose-free Earle’s solution (OGD medium) [116.4 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 0.8 mmol/L MgSO4, 2.6 mmol/L NaH2PO4, 26.2 mmol/L NaHCO3, and 20.1 mmol/L HEPES (pH 7.4)] to remove glucose contained in the culture medium, and incubated in adequate volume OGD medium in an anaerobic chamber containing nitrogen (95%) and carbon dioxide (5%) mixture for the various indicated times. Then the OGD medium was replaced to complete culture medium with a final concentration of 4.5 mg/L glucose, followed by incubation for 24 h (OGD-Rep) in normal conditions. Control cells were incubated in glucose-containing (5 mM) Earle’s solution (control medium) at 37°C in a regular CO2 (5%) incubator (normoxic condition).
Assays of cell survival and cell damage
Cell survival was evaluated by MTT assay. Neuronal cells (5×104) were cultured in a 96-well plate. After 6-8 days in culture or reperfusion, 10 μl of assay reagent MTT (Sigma, MO, USA) (5 mg/ml) was added and incubated at 37°C for 4 h. Then the culture medium were removed carefully and 100 μl DMSO was added for 10 min at 37°C. The absorbance at 570 nm was assayed by an ELISA micro-plate reader (Molecular Devices Inc, USA). The cell injury was assessed by a quantitative measurement of released LDH. Reperfusion 24 hours after exposure to anoxia and non-glucose environment, cells were incubated in complete culture medium containing 1% Triton X-100 (pH 7.4) at 37°C for 30 min. The absorbance at 340 nm was assayed by an ELISA micro-plate reader (Molecular Devices Inc, USA) every 30 s for 3 min.
Electrophysiology recording
The procedure for whole-cell patch-clamp recording was described in our previous study [22] with HEKA EPC-10 patch clamp amplifier (HEKA, Germany). The signals were recorded and analyzed with Pulse + pulsefit software 8.6 (HEKA, Germany). Borosilicate glass pipettes (1.5 mm diameter) were pulled with a two-stage microelectrodes puller (Narrishige, Japan), and the resistance of the recording electrode was 2-5 MΩ. To record ASIC current, cortical neurons tested were voltage-clamped at -80 mV throughout the experiments. A multibarrel perfusion system was used as our previously research to achieve a rapid exchange of extracellular solutions (150 mM NaCl, 5 mM KCl, 1 mM MgCl2·6 H2O, 2 mM CaCl2, 10 mM Glucose, 10 mM HEPES, pH 7.4 or 6 with Tris-OH). The pipette solution was as follows: Record L-VGCC current with solution contained: 110 mM NaCl, 5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 11 mM glucose, 10 mM HEPES, 5 mM 4-AP, 25 mM TEA-Cl and 1 μM tetrodotoxin. Recording pipettes were filled with the following solution: 64 mM CsF, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM EGTA, 10 mM HEPES, and 5 mM Tris-ATP. To isolate the voltage-dependent calcium currents, tetrodotoxin (1 μM), 4-AP (5 mM) and TEA-Cl (25 mM) were added in the bath solution. The current signals were sampled by 10 kHz and filtered at 3 kHz.
Calcium imaging
For [Ca2+]i measurements, cultured neurons were washed with standard ECF (150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Glucose, 10 mM HEPES, pH 7.4 with Tris-OH) and loaded with 1 μmol/L Fura2/AM (Molecular Probes) with 0.1% DMSO and 1% BSA in ECF for 25 min at 37°C. The cover slips were then transferred to a chamber mounted on the movable stage of an inverted microscope (Olympus IX-70, Japan). The ratio of Fura-2 signals at 340 and 380 nm was obtained. Data were digitized at 1-s intervals. Recording and image analysis were performed by using TILLVISION 4.0 software (Universal Imaging, Media, PA). The values were exported to Sigmaplot 8.0 for further analysis and [Ca2+] response amplitude was defined as the peak of ΔF/F.
Western-blotting
Cortical neurons were collected and lysed with 100 μl lyses buffer (50 mM Tris-HCl, pH 7.4, 10 mM EDTA, 100 mM NaCl, 1% Nonidet P-40, 20 mM NaF, 3 mM Na3VO4, 1 mM PMSF). The lysates were centrifuged and prepared for blotting. A 50 μg of protein aliquot were loaded and was subjected to 10% SDS-PAGE electrophoresis for 1 h. The proteins were then transferred to nitrocellulose membranes using a transfer system (Bio-Rad, CA, USA). The membranes were blocked and incubation with anti-ASIC1 (Alomone labs, Israel) or anit-ASIC2a (Alomone labs, Israel), Following washing, peroxidase-coupled secondary antibody (anti-rabbit IgG) was added and incubated for 1 h. The membrane was washed and specific bands were developed on films using the enhanced chemiluminescence technique (SuperSignal West Pico; Pierce, USA), the intensity of the bands was measured using IPP image software. The total protein content was normalized using mouse β-tubulin (Cell Signaling Technology, MA, USA) antibodies.
Immunofluorescence measurement
Cerebral cortical neurons were collected and fixed with 4% paraformaldehyde in PBS for 30 min. Then permeabilized with PBS/0.3% Triton X-100 for 30 min, and all cells were blocked with 2% goat serum and 1% bovine serum albumin (BSA) in PBS for 1 h, and then incubated with anti-ASIC1 (Alomone labs, Israel) or anti-ASIC2a (Alomone labs, Israel) overnight at 4°C. Cells were rinsed in PBS three times (10 min each time), and then incubated with anti-rabbit tetramethyl rhodamine isothiocyanate antibody (Pierce) for 1 h at room temperature. Fluorescence density was collected by Olympus IX-70 microscope and assayed with IPP software.
Statistical analysis
All experimental data are presented as means ± SEM. Differences between groups were analyzed by one-way ANOVA followed by post hoc Tukey’s test. A value of P<0.05 was considered statistically significant.
Results
PICK1-knockout protects primary cultured mouse cortical neurons from OGD-Rep insults
To mimic in vivo oxygen glucose deprivation condition and imperative reperfusion situation in brain ischemia and repairing process, cultured cortical neurons were subjected to OGD-Rep in vitro. OGD-Rep did compromise the cell survival as analyzed by trypan blue staining, which showed an obvious decrease of neuron numbers, and also decreased the metabolic viabilities of cells as demonstrated by MTT assay (Figure 1A). More interesting, as compared with wild-type neurons, the heterozygote neurons and knockout neurons showed a much more resistant property to ischemic damages, especially the knockout neurons. These results were parallel to the results of trypan blue staining (data not shown).
Figure 1.
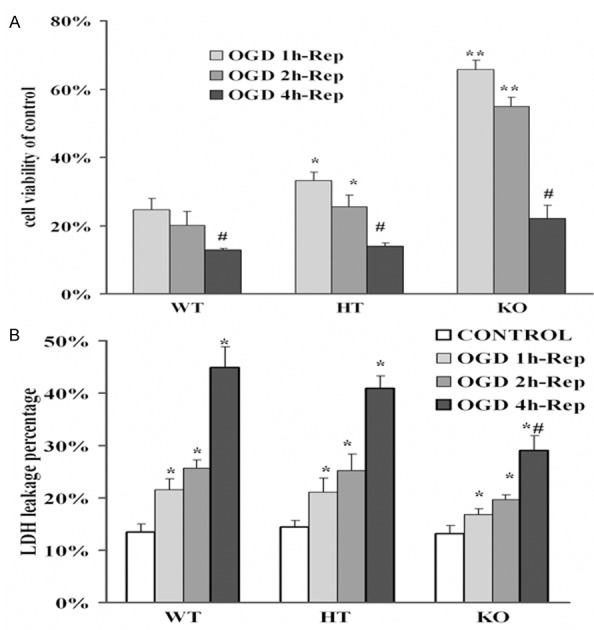
The cells viability and LDH released in neuronal cells after OGD-Rep. Cells were prepared and under OGD-Rep treatment for 24 h, then collected cells for MTT assay and LDH release test. A. Cell viability was tested by MTT assay subjected to different time courses of OGD treatment and reperfusion 24 hours compared with sham cells (cell viability =100%). B. LDH leak percentages of cells subjected to different time courses of OGD-Rep. Data are the mean ± SD of 5 well per group. *P<0.05 vs wild type group, **P<0.01 vs wild type group, # implies P<0.01 vs OGD 1 h or OGD 2 h-Rep of each cell-type. WT: Wild type cells; HT: Heterozygote cells; KO: PICK1 knockout cells.
To further study the effects of OGD-Rep on the necrotic cell injuries of cortical neurons, we analyzed the LDH leakages to reflect the complexity of the cellular membrane. Figure 1B represents all ischemic neurons showed a time-dependent increasing of LDH leak as compared with non-ischemic cells. Both heterozygote neurons and knockout ones also showed a neuropretective effect against OGD-Rep, especially in the knockout cells.
PICK1-knockout decreases ASIC1 and ASIC2a protein expression in primary cultured mouse cortical neurons during OGD-Rep
To test the protein abundances of ASIC1 and ASIC2a in neurons during OGD-Rep, the cells were prepared for western blot and immunofluorescence analysis. As shown in Figure 2A and 2C, before OGD, PICK1-knockout neurons showed a significant reduction in the expression of ASIC1 as compared with wild-type neurons. However, OGD-Rep increased the AISC1 expression in wild-type and heterozygote cells, but dramatically decreased the AISC1 protein in knockout neurons (P<0.05). The protein abundance of ASIC2a showed similar trend as AISC1 in these three type cells during OGD-Rep (Figure 2A and 2C).
Figure 2.

ASIC1 and ASIC2a protein expression during OGD-Rep in three types of neurons. Cells were treated with OGD-Rep and prepared for immunofluorescence and western blot analysis. A, C. Representative pictures and summary data showing expressions of ASIC1 and ASIC2a using immunofluorescence. B, D. Representative bands and summary data of ASIC1 and ASIC2a expression in western blotting. Data are the mean ± SD in each group. *P<0.05 vs wild type group, **P<0.01 vs wild type group, # implies P<0.01 vs OGD 1 h or OGD 2 h-Rep of each cell-type. WT: Wild type cells; HT: Heterozygote cells; KO: PICK1 knockout cells.
PICK1-knockout down-regulates activation of whole cell ASIC current during OGD-Rep
Applying whole cell patch clamp mode, with pH 6.0 fast acidification and voltage clamped at -80 mv, three types of ASICs currents were recorded in cortical neurons (Figure 3A). Majority cells (84.2%) presented a rapid activation and desensitized within 2000±186 ms (Type I), and 13.2% cells presented a rapid activation and a quicker desensitization within 300±60 ms (Type II). Both Type I and Type II decayed along with the acid pulses (Figure 3B). Eighty-two neurons (79.3%) showed a tachyphylaxis to continual extracellular acid applications. The I-V curve and the amiloride-blockable characteristics both confirmed the participation of ASICs (Figure 3C).
Figure 3.

Whole cell ASICs currents recorded in primary cultured cortical neurons due to OGD-Rep and PICK1 knockout. Cells were treated with OGD-Rep and collected for whole-cell patch-clamp recording. A. Three types of ASIC current with different activation and desensitization time constants were recorded, the majority presented the left pattern. B. Two kinds of responses to continual acid applications. C. ASIC current presented a linear I-V curve and a blockable effect to 100 μM amiloride. D. Mean current amplitude of cell after OGD-Rep. E. Representative traces of modifications of ASICs current amplitude in OGD-Rep neurons in each cell-type. F. Summary data of modifications of ASICs current density in OGD-Rep neurons in each cell-type. Data are the mean ± SD in each group. *P<0.05 vs wild-type control group, #P<0.05 vs wild-type OGD-Rep groups. WT: Wild type cells; HT: Heterozygote cells; KO: PICK1 knockout cells.
As shown in Figure 3D and 3E, both the amplitude and density of ASICs currents after OGD-Rep increased in wild-type neurons, but decreased in knockout neurons. However, it has no effect on heterozygote neurons. PICK1-knockout cells also showed a lower ASIC current than wild-type or heterozygote neurons in control conditions (P<0.05 vs. wild-type and vs. heterozygote, Figure 3F). Other electrophysiological and pharmacological characteristics of ASICs currents such as kinetics, permeation properties and amiloride-blockade were not altered by OGD-Rep (data not shown).
PICK1-knockout suppressed intracellular calcium transients induced by acid applications after OGD-Rep
Majority neurons presented a relatively stable inward current at the third or the fourth pulse, so the later currents we analyzed were all from the fourth pulse when the currents ceased decaying (Figure 4A). Blocked calcium transient by 100 μM amiloride completely and reversibly, there was no transient in Ca2+-free ECF mediated by acid, but an obvious calcium transient Ca2+-containing ECF. Incubated with 1 μM thapsigargin (TG) to deplete the intracellular calcium store, the transient was just 10% less than untreated one before neurons bathed in Ca2+-containing ECF (Figure 4B). Above results demonstrated the intracellular Ca2+ increase mainly comes from extracellular Ca2+ influx, and calcium induced calcium release (CICR) also plays partial roles.
Figure 4.
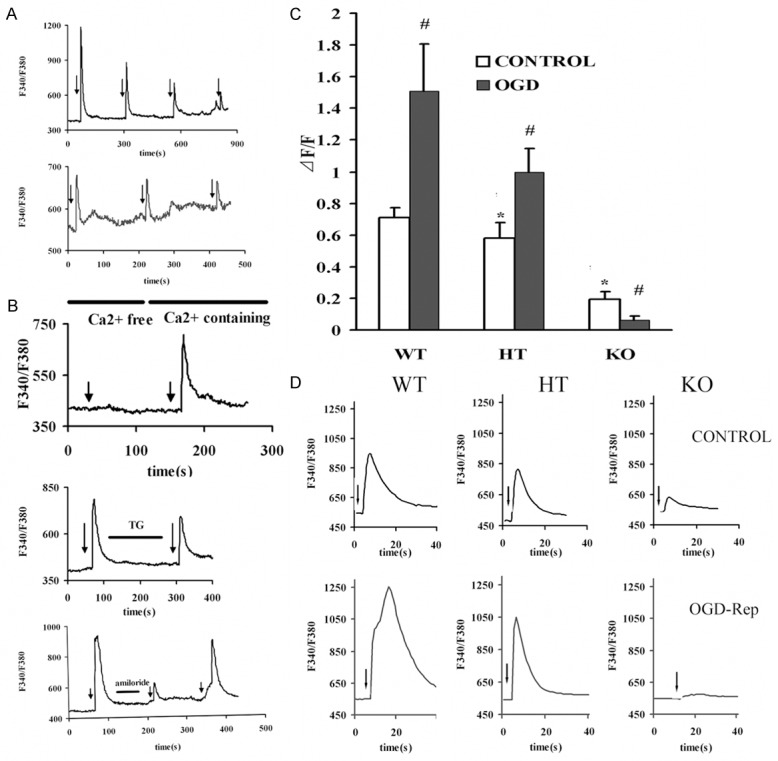
Intracellular calcium transients mediated by pH 6.0 fast applications due to OGD-Rep and PICK1 knockout. Cells were treated with OGD-Rep and collected for Calcium imaging. A. Two kinds of decay pattern were observed. The upper kind showed gentle decays and stable transient at the fourth acid administration, the lower presented dramatic decays and no stable transient could be attained. B. The amiloride-blockable characteristic and source of calcium transients mediated by acidosis. C. Summary data of alterations of mean calcium transients mediated by acidosis in OGD-Rep neurons in each cell-type. D. Representative traces showing 123% increase in wile-type, 70.7% increase in heterozygote and 68.4% decrease in knockout neurons following OGD-Rep. Data are the mean ± SD in each group. The arrows indicated pH 6.0 application. *P<0.05 vs wild-type control group, #P<0.05 vs control neurons of each group. WT: Wild type cells; HT: Heterozygote cells; KO: PICK1 knockout cells.
Paralleling to ASICs currents, both the peak of [Ca2+]i (Δ[Ca2+]i) and the normalized Δ[Ca2+]i (ΔF/F) were altered. As in Figure 4C and 4D, the acid induced normalized Δ[Ca2+]i (ΔF/F) augmented greatly after OGD-Rep in wild-type neurons, from 0.71±0.06 to 1.51±0.30 (P<0.05, wild-type vs. control); on the contrary, in knockout neurons it was notably suppressed (68.4%). In the heterozygote neurons, the calcium transient was also increased after OGD (P<0.05, heterozygote vs. control). Before OGD, heterozygote and PICK1-knockout neurons showed smaller calcium transients than wild-type. After OGD-Rep, the differences among groups were more dramatic. PICK1-knockout reduced ASIC current density and calcium transient mediated by acidosis.
PICK1-knockout reduces L-VGCC density during OGD-Rep
Although the roles of L-VGCC ((L-type voltage gated calcium channel)) and NMDA receptor in brain ischemia have been challenged, they still played a fractional part, so we also observed the modifications of L-VGCC. As in Figure 5A and 5B, we found a dramatic increase of current density in all neurons after OGD-Rep, but the elevation degree of knockout neurons (100%) was significantly reduced than wild-type (158%, P<0.05) or heterozygote cells (148%, P<0.05). These data suggest that in the PICK1-knockout, protection of brain ischemia-reperfusion process not only involves ASICs, but also relates with other factors such as apoptosis and L-VGCC activation.
Figure 5.
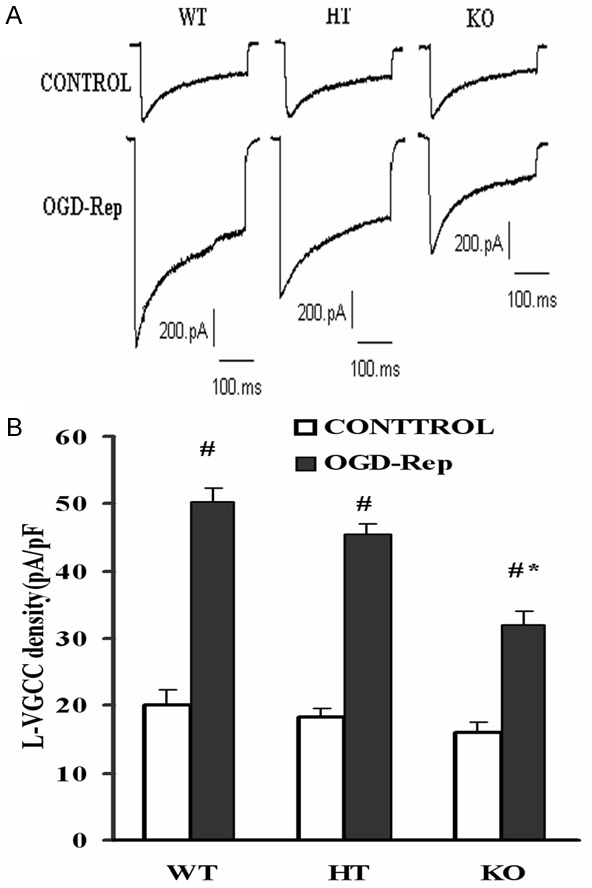
L-VGCC activation after OGD-Rep in three types of cortical neurons. Cells were treated with OGD-Rep and collected for whole-cell patch-clamp recording. A. Typical L-VGCC current recorded in three types of neurons before and after OGD-Rep. B. Analytical plot showing a dramatic increase of current density in all neurons after OGD-Rep. Data are the mean ± SD in each group. #P<0.05 vs control, *P<0.05 vs corresponding wild-type or heterozygote group. WT: Wild type cells; HT: Heterozygote cells; KO: PICK1 knockout cells.
Discussion
Ischemic brain injury is resulted from complicated cellular mechanisms. Reperfusion after cerebral ischemia can cause serious reperfusion injury, and acidosis is a common characteristic of brain damage [23]. In this study, OGD-Rep significantly reduced the neural cells viability with time dependent way. In the meantime, the release of LDH was dramatically increased with OGD-Rep. These results are similar with previous studies that ODG-Rep induced the neural cells damage. Acidosis is a common characteristic of brain damage following cerebral ischemia reperfusion injury. It is well known that ischemia results in a marked reduction in tissue pH, and that acidosis is an important determinant of neurological injury [10]. In the ischemic brain, pH falls to 6.0 due to the accumulation of lactic acid and as a consequence of protons produced by adenosine triphosphate hydrolysis [24]. Increase in glycolysis and lactate production under hypoxic conditions result in acidosis, which is the most important characteristic of hypoxia [25].
This is the well-known NMDA (N-methyl-D-aspartate)-medated excitotoxicity mechanism that contributes to the cerebral ischemia-induced neuronal injury [26]. In addition to NMDA-dependent mechanisms, recent studies have shown that acid-sensing ion channel 1a (ASIC1a) contributes to neuronal injury after ischemia and reperfusion [7,9]. Acidosis can activate homomeric ASIC1a, causing a large influx of Na+ and Ca2+, leading to neuronal injury [27]. Agents that potentiate ASIC1a activity and expression have been shown to exacerbate acidosis-mediated neuronal injury and/or ischemic outcomes [28,29]. In this OGD-Rep model, OGD-Rep increased the expression of ASIC1 and ASIC2a induced by acidosis, which will exacerbate neuronal injury.
In most CNS neurons, lowering the extracellular pH in an ischemic brain induces membrane depolarization and an increase in intracellular Ca2+ [12]. ASICs are voltage-independent, proton-gated cation-selective channels mostly permeable to Na+ ion. Among all the ASICs, homomeric ASIC1a and heteromeric ASIC1a/2b channels are known to have permeability to Ca2+. Activation of these channels can cause accumulation of intracellular Ca2+ concentration in neurons [11,12]. Similarly with previous studies, ASICs currents we recorded in mouse cortical neurons also presented discrepant desensitization constants and showed two different tachyphylaxis patterns. Homomeric ASIC1a channels had a relative high sensitivity to H+ with the pH of half maximal activation (pH0.5) of 6.2 [30], while the pH0.5 of homomeric ASIC2a channels was 4.4 [31]. ASICs currents recorded in our experiment were elicited by pH 6.0, and so ASIC1a subunit contributed more to the inward currents composition. OGD treatment induced a moderate increase of the amplitude of ASIC currents and a dramatic decrease in ASIC desensitization [27]. Similarly with the above observations, the amplitude of ASIC currents increased after OGD-Rep in wild-type cortical neurons, but different from their findings, we observed no changes of desensitization constants after the ischemia-reperfusion process. This difference may come from the different models we used, they used OGD 1 hour (a mere ischemia injury model), and we applied OGD 4 hours and then reperfusion 24 hours (an ischemia-reperfusion injury model).
ASIC1a, the major ASIC subunit with Ca2+ permeability, is highly expressed in neurons [11,12]. Ca2+ imaging in COS-7 cells transiently expressing ASIC1a recently demonstrated that homomeric ASIC1a channels are a major non-voltage-gated pathway for Ca2+ entry in cells [12]. We found that 70% mouse cortical neurons showed a calcium transient to acid pH 6.0 application, that is to say, most of the cortical neurons are homomeric ASIC1a. The rest 30% neurons may be ASIC1/ASIC2a heteromeric or ASIC2a homomeric. The calcium transients also decayed along with continual acid application like homomeric ASIC1a currents. These findings further indicate different ASIC subunit composition exists in neurons, and ASIC1a is still the predominant functional subunit in brain. We firstly found that the extent of calcium transient mediated by acid dramatically elevated after OGD-Rep in wild-type neurons. In all, over-activation of ASICs during ischemia-reperfusion permeates more Na+ and Ca2+ to evoke depolarization of cellular membrane even trains of action potential that facilitates activation of NMDA receptor and induces intracellular Ca2+ overload, which deteriorates brain ischemia process where ASICs play an incredible important part.
ASICs have two transmembrane domains separated by a large extracellular loop, with cytoplasmic amino and C termini [32]. In sensory neurons and in the brain, the PDZ domain-containing protein PICK-1 was shown to interact with the C termini of both ASIC1 and ASIC2 [33]. It has been demonstrated that PICK-1 actually serves as an intermediary protein through which protein kinase C acts to up-regulate ASIC2a expression [34] and to modulate heteromultimeric ASIC2b-ASIC3 channel expression and pH sensitivity [33]. Cerebral ischemia up-regulated the expression of various PKC subunits including PKCα [35], PKCα would interact with its binding protein PICK1 to up-regulate the expression and re-distribution of ASICs. In all, functional upregulation of ASICs plays vital roles in brain ischemia-reperfusion, especially Ca2+-permeable ASIC1a.
In most of the cases, the binding of PICK1 regulates the trafficking of its binding partners by altering either their subcellular targetings or surface expressions [15], but for ASICs here, PICK1 acted not only as an interacting protein but also as an important regulation factor to modulate the post-transcriptional process of ASICs in ischemia process. In our present study, PICK1-knockout showed a decreased ASICs function and protein expression but equal gene expression to wild-type neurons induced by OGD-Rep. By PICK1 knockout cells, the ASICs protein expression was significantly reduced. In the meantime, ASICs current and the calcium transients were decreased by PICK1 knockout. These results indicated that PICK1 knockout rescued the neuronal cells injury by acidosis induced ASICs activation after OGD-Rep. Interestingly, we firstly found besides ASICs, L-VGCC may be relatively down-regulated by disrupting PICK1 gene, which also played proportional parts in neuroprotective effect of PICK1-knockout in brain ischemia. It further indicated that PICK1 may probably be a key upstream regulator in brain ischemia process.
Present study shows that ASICs over-activation and over-expression are responsible for neuronal injury during ischemia-reperfusion insults in primary cultured mouse cortical neurons. Depleting PICK1 gene showed an obvious neuroprotective effect in acidosis condition induced by OGD-Rep, which was related with suppressed activation of ASICs and inhibited ASICs current and the calcium transients. PICK1 gene might be a new target to prevent and treat acidosis related diseases such as brain ischemia and epilepsy.
Acknowledgements
This work is supported by grants from the National Science Foundation of China (NSFC No. 81202539) to Dr. Jin Cheng and NSFC No. 81401030 to Dr. Xihong Ye.
Disclosure of conflict of interest
None.
References
- 1.Kim KW, Jin Y. Neuronal responses to stress and injury in C. elegans. FEBS Lett. 2015;589:1644–1652. doi: 10.1016/j.febslet.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seta KA, Yuan Y, Spicer Z, Lu G, Bedard J, Ferguson TK, Pathrose P, Cole-Strauss A, Kaufhold A, Millhorn DE. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium. 2004;36:331–340. doi: 10.1016/j.ceca.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 4.McDonald JW, Bhattacharyya T, Sensi SL, Lobner D, Ying HS, Canzoniero LM, Choi DW. Extracellular acidity potentiates AMPA receptor-mediated cortical neuronal death. J Neurosci. 1998;18:6290–6299. doi: 10.1523/JNEUROSCI.18-16-06290.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benveniste M, Dingledine R. Limiting stroke-induced damage by targeting an acid channel. N Engl J Med. 2005;352:85–86. doi: 10.1056/NEJMcibr045010. [DOI] [PubMed] [Google Scholar]
- 6.Xiong ZG, Chu XP, Simon RP. Acid sensing ion channels--novel therapeutic targets for ischemic brain injury. Front Biosci. 2007;12:1376–1386. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- 7.Osmakov DI, Andreev YA, Kozlov SA. Acid-sensing ion channels and their modulators. Biochemistry (Mosc) 2014;79:1528–1545. doi: 10.1134/S0006297914130069. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez de la Rosa D, Krueger SR, Kolar A, Shao D, Fitzsimonds RM, Canessa CM. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol. 2003;546:77–87. doi: 10.1113/jphysiol.2002.030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu XP, Grasing KA, Wang JQ. Acid-sensing ion channels contribute to neurotoxicity. Transl Stroke Res. 2014;5:69–78. doi: 10.1007/s12975-013-0305-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uria-Avellanal C, Robertson NJ. Na(+)/H(+) exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res. 2014;5:79–98. doi: 10.1007/s12975-013-0322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherwood TW, Lee KG, Gormley MG, Askwith CC. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci. 2011;31:9723–9734. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeh PW, Kress M. Molecular physiology of proton transduction in nociceptors. Curr Opin Pharmacol. 2001;1:45–51. doi: 10.1016/s1471-4892(01)00014-5. [DOI] [PubMed] [Google Scholar]
- 14.Johnson MB, Jin K, Minami M, Chen D, Simon RP. Global ischemia induces expression of acid-sensing ion channel 2a in rat brain. J Cereb Blood Flow Metab. 2001;21:734–740. doi: 10.1097/00004647-200106000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Xia J. Structure and function of PICK1. Neurosignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- 16.Hruska-Hageman AM, Wemmie JA, Price MP, Welsh MJ. Interaction of the synaptic protein PICK1 (protein interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain Na+ channel 1) and ASIC (acid-sensing ion channel) Biochem J. 2002;361:443–450. doi: 10.1042/0264-6021:3610443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. J Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin W, Shen C, Jing L, Zha XM, Xia J. PICK1 regulates the trafficking of ASIC1a and acidotoxicity in a BAR domain lipid binding-dependent manner. Mol Brain. 2010;3:39. doi: 10.1186/1756-6606-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu ZL, Huang C, Fu H, Jin Y, Wu WN, Xiong QJ, Xie N, Long LH, Chen JG, Wang F. Disruption of PICK1 attenuates the function of ASICs and PKC regulation of ASICs. Am J Physiol Cell Physiol. 2010;299:C1355–1362. doi: 10.1152/ajpcell.00569.2009. [DOI] [PubMed] [Google Scholar]
- 21.Ming Y, Zhang H, Long L, Wang F, Chen J, Zhen X. Modulation of Ca2+ signals by phosphatidylinositol-linked novel D1 dopamine receptor in hippocampal neurons. J Neurochem. 2006;98:1316–1323. doi: 10.1111/j.1471-4159.2006.03961.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Daggett H, De Waard M, Heinemann SH, Hoshi T. Nitric oxide augments voltage-gated P/Q-type Ca (2+) channels constituting a putative positive feedback loop. Free Radic Biol Med. 2002;32:638–649. doi: 10.1016/s0891-5849(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 23.Simon R, Xiong Z. Acidotoxicity in brain ischaemia. Biochem Soc Trans. 2006;34:1356–1361. doi: 10.1042/BST0341356. [DOI] [PubMed] [Google Scholar]
- 24.Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260:R581–588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JW, Gao P, Dang CV. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev. 2007;26:291–298. doi: 10.1007/s10555-007-9060-4. [DOI] [PubMed] [Google Scholar]
- 26.Lai TW, Shyu WC, Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med. 2011;17:266–275. doi: 10.1016/j.molmed.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 28.Duan B, Wang YZ, Yang T, Chu XP, Yu Y, Huang Y, Cao H, Hansen J, Simon RP, Zhu MX, Xiong ZG, Xu TL. Extracellular spermine exacerbates ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis. J Neurosci. 2011;31:2101–2112. doi: 10.1523/JNEUROSCI.4351-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng WZ, Liu DS, Duan B, Song XL, Wang X, Wei D, Jiang W, Zhu MX, Li Y, Xu TL. Molecular mechanism of constitutive endocytosis of Acid-sensing ion channel 1a and its protective function in acidosis-induced neuronal death. J Neurosci. 2013;33:7066–7078. doi: 10.1523/JNEUROSCI.5206-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldmann R, Lazdunski M. H (+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- 31.Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB, McNaughton PA. Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci. 2007;27:13251–13260. doi: 10.1523/JNEUROSCI.2135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog Neurobiol. 2014;115:189–209. doi: 10.1016/j.pneurobio.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deval E, Salinas M, Baron A, Lingueglia E, Lazdunski M. ASIC2b-dependent regulation of ASIC3, an essential acid-sensing ion channel subunit in sensory neurons via the partner protein PICK-1. J Biol Chem. 2004;279:19531–19539. doi: 10.1074/jbc.M313078200. [DOI] [PubMed] [Google Scholar]
- 34.Baron A, Deval E, Salinas M, Lingueglia E, Voilley N, Lazdunski M. Protein kinase C stimulates the acid-sensing ion channel ASIC2a via the PDZ domain-containing protein PICK1. J Biol Chem. 2002;277:50463–50468. doi: 10.1074/jbc.M208848200. [DOI] [PubMed] [Google Scholar]
- 35.Harada K, Maekawa T, Abu Shama KM, Yamashima T, Yoshida K. Translocation and down-regulation of protein kinase C-alpha, -beta, and -gamma isoforms during ischemia-reperfusion in rat brain. J Neurochem. 1999;72:2556–2564. doi: 10.1046/j.1471-4159.1999.0722556.x. [DOI] [PubMed] [Google Scholar]