

Abstract
Ischemia-reperfusion (I/R) injury can lead to apoptotic death of heart cells and subsequently heart failure. Propranolol is widely used in the management of cardiovascular disorders, but the mechanism is still unclear. Our previous studies showed that activated protein kinase C1 (RACK1) was significantly down-regulated in human umbilical vein endothelial cells by S-propranolol. RACK1 may be a target protein of S-propranolol during I/R. At present, we constructed a lentiviral expression vector for RNA interference (RNAi) of RACK1. The interference efficiency of the lentivirus was confirmed by RT-PCR and western blot. H9C2 cells infected with Lv-RACK1-shRNA or control were subjected to simulate I/R in the presence and absence of S-propranolol. The release of cytokines and chemokines was determined by ELISA assay. Flow cytometry was employed to determine mitochondrial membrane potential (MMP), Ca2+ concentration, reactive oxygen species (ROS) production, and cell apoptosis. We found that RACK1 RNAi and S-propranolol treatment remarkably protected I/R injured cells from apoptosis via attenuating the release of cytokines and chemokines, Ca2+ overload, ROS concentration, and MMP. Furthermore, RACK1 RNAi and S-propranolol, separately and in combination, significantly reduced caspase-3 activity, cytochrome c release and JNK activation. RACK 1 can be considered as a target for drug development.
Keywords: Myocardial ischemia/reperfusion injury, RACK1, propranolol, MAPK, oxidative stress, apoptosis
Introduction
Cardiovascular diseases represent one of the main causes of death in the world. High-fat diet, a sedentary way of life and genetic factors seem to account for the development of cardiovascular diseases including atherosclerosis and heart stroke. Ischemic damage, following coronary occlusion, occurs not only in the myocardial cells, but in the capillaries, venules, and arterioles supplying the myocardium as well [1]. Both ischemia and reperfusion contribute to cell and tissue damage after cardiac infarction. Myocardial ischemia-reperfusion (I/R) initiates mal-distribution of ions and various signaling mechanisms, leading to oxidative injury and inflammatory responses that include liberation of cytokines [2] and free radicals [3], up- and down-regulation of various genes and their proteins [4,5] and cell death by apoptosis [6] and/or necrosis [7]. It is also well known that reactive oxygen species (ROS), mediators of the oxidative stress, play deleterious effects on vasculature and myocardium.
Propranolol selectively blocks beta-adrenergic receptors [8]. This action has provided a new and potent approach to the therapy of a variety of cardiovascular disorders. It has proved particularly effective in treatment of the symptoms of angina pectoris [9]. It produced a decrease in externally measured indices of myocardial mechanical effort and consequently a fall in myocardial oxygen demands in male patients [10]. It also has been shown to reduce the extent of myocardial infarction, and decrease microvascular ischemic injury [11]. Propranolol not only protects the ischemic myocardial cells, but also significantly decreases the ischemic microvascular changes [12]. However, the mechanism of propranolol in the treatment of myocardial damage is still unclear.
In our previous proteomics study, we found that receptor of activated protein kinase C1 (RACK1, also called guanine nucleotide-binding protein subunit beta- 2 like 1 (GBLP, Swiss-Prot entry P63244) is significantly down-regulated in human umbilical vein endothelial cell treated with R (+) or S (-)-propranolol. S (-)-propranolol has more significant effect on the downregulation of RACK1 than R (+) propranolol, which indicated that the stereochemical structure of propranolol may be selectively related with prevention effect on myocardial damage. RACK-1 regulates several key processes that involve contact with the extracellular matrix, such as cell spreading, the establishment of focal adhesion and cell-cell contacts. RACK1 proteins were validated as bona fide interacting partners of ubinuclein (Ubn-1), a novel nuclear and adherent junction complex components protein interacting with cellular and viral transcription factors. In the context of Epstein-Barr virus (EBV, has been linked to a decrease in the production of proinflammatory mediators as well as an impairment of phagocytosis) infection, interaction of the viral transcription factor EB1 with RACK-1 could result in inhibition of protein kinases C (PKC) activity, which affects functions of EBV-infected monocytes such as phagocytosis. PKC activated by signals such as increases in concentration of Ca2+, RACK-1 has been described to interact with the integrin receptor and interferes with integrin signaling pathways by recruiting specific kinases. RACK-1 interacts also with spectrin, IP3R (receptor for inositol 1, 4, 5 triphosphate) and Scp160p which is a member of the vigilin family of RNA-binding proteins. Interestingly these partners of RACK-1, including integrin, was also identified in this study as interacting partners of Ubn-1 [13]. All the biofunction of RACK1 may have critical effect on mycardic cells.
RACK1 is considered as the key protein related with myocardial damage. In present study, we construct RNA interference (RNAi) model, an RNA-dependent gene silencing process initiated by the double-stranded RNA (dsRNA) [14]. RACK1 expression will be down-regulated in ischemia/reperfusion H9C2 cells, we characterized RACK1 is critical for process of myocardial damage and the mechanism of S-propranolol and R-propranolol effect on the I/R injury.
Materials and methods
Reagents
S-propranolol and R-propranolol (Sigma), Trizol (Invitrogen, 15596-026), DEPC water (JRDUN Biotechnogy BYL40387), SYBR Green PCR kit and Transcription Kit (Thermo, F-415XL) for PCR analysis.
Dulbecco’s Modified Eagle’s Medium (DMEM) and fetal bovine serums (FBS) were purchased from Gibco. H2O2 was obtained from Fisher Scientific (USA). N-acetyl-L-cysteine (NAC, purity > 99%) were from sigma, Kits for iNOS (Nanjing, China), antibodies [anti-RACK1 (No.ab62735), anti-p-JNK1/2, anti-JNK1/2, anti-Cytc, anti-Caspase3, anti-Bcl-2 and anti-GAPDH were used for western blot analysis.
Cell culture and treatment
The H9C2 cell, a rat cardiac myoblasts cell line, was supplied by Sun Yat-Sen University Experimental Animal Center (Guangzhou, China). H9C2 cells were cultured in DMEM supplemented with 10% FBS at 37°C in an atmosphere of 5% CO2 and 95% air. I/R of H9C2 rat embryonic cardiac myoblasts was achieved by culturing the cells in low glucose DMEM without FBS in a hypoxia chamber, saturated with 5% CO2 for 24 h and following.
Assay of apoptosis
To detect early stages of apoptosis, an Annexin-VFLUOS staining kit (Roche) was used according to the manufacturer’s instructions. Briefly, I/R cells were washed with PBS and incubated with Annexin-V/propidium iodide (PI)/ Hoechst 33342 for 15 min at room temperature. Cells were visualized with a fluorescence microscope and Annexin-V positive and PI negative cells were assessed as apoptotic. The percentage of apoptotic cells was calculated by dividing Annexin-V positive cells by the total number of cells visualized by Hoechst staining. Three randomly chosen optical fields per preparation were digitized and each experimental condition was done in triplicate. The mean percentage of apoptotic cells was averaged over a total of three independent experiments.
Measurement of the mitochondrial membrane potential
The mitochondrial membrane potential (MMP) was assessed using a fluorescent dye, Rhodamine (Rh123), a cell-permeable carionic dye that preferentially enters mitochondria based on the highly negative MMP. Depolarization of MMP results in a loss of MMP from the mitochondria and a decrease in green fluorescence. H9c2 cells were cultured in a slide with EMEM-F12. After the indicated treatments, the slides were washed two times with PBS. The I/R cells were incubated with 1 mg/l Rh123 at 37°C for 30 min. in the incubator, washed briefly with PBS three times and air-dried again. Then, fluorescence was measured over the hold field of vision by using a fluorescent microscope connected to an imaging system (BX50-FLA, Olympus).
The MFI of Rh123 from five random fields was analysed using the Image J 1.41o software, the MFI was taken as an index of the levels of MMP. The experiment was carried out in triplicate.
Assay of intracellular Ca2+ mobilization
After loading with Fluo-3, the cells were pre-incubated for 15 min at room temperature with different concentrations of the CXCR4 antagonist, AMD3100, diluted in Ca2+ flux assay buffer. For each sample, a 20-s baseline monitoring was performed in the flow cytometer. Then sample aspiration was briefly paused and SDF-1 was quickly added, and then the Ca2+ response was measured as the change in green fluorescence intensity of the cells as a function of time. Analysis was performed with the FAC Scanflow cytometer (Becton Dickinson) with an air-cooled 488-nm argonion laser (Spectra Physics) and CellQuest software (Becton Dickinson).
Measurement of intracellular ROS
Intracellular ROS levels were examined using 2’,7’-dichlorofluorescein diacetat (DCFH-DA) (Molecular Probes). Osteoblasts (3 × 105 cells/well in 6-well plates) were treated with hydrogen peroxide in the presence or absence of test compounds or NAC (antioxidant) and incubated for 48 h, pre-incubated with various concentrations of curculigoside and NAC for 2 h, and incubated with 400 µM H2O2 for 48 h. The supernatant was discarded and cells were washed with PBS, added with 5 µM DCFH-DA, and incubated for 30 min at 37°C in the dark. Excessive DCFH-DA was removed. Cells were washed twice with PBS, collected and analyzed for fluorescence of 2’,7’-dichlorofluorescein (DCF) at 485 nm excitation and at 525 nm emission by flow cytometry.
TNF-α, IL6, IL-8 and IL-1b detection in conditioned media
A commercial ELISA assay (R&D Systems, Minneapolis, MN, USA) was used to measure the protein level of rat TNF-α, IL6, IL-8 and IL-1b in conditioned media after S-propranolol and R-propranolol treatment (0-500 ng/mL), as per the manufacturer’s instructions.
Analysis of antioxidant enzymes
H9C2 (2 × 106 cells/100 mm culture dish) were seeded and treated as before. At the end of incubation, the medium in the 100 mm dish was discarded. H9C2 cells were washed with ice-cold PBS three times, collected, centrifuged at 1500 × g for 5 min, homogenated in PBS by sonicating 10 times for 5 s interval 5 s, and centrifuged at 12000 × g for 10 min at 4°C. The activity of antioxidant enzymes (iNOS) in the supernatant was determined according to the guidelines of the kits (Nanjing Jiancheng Bioengineering Institute, China). The values were expressed as units per mg protein and the protein concentration was determined with a BCA protein assay kit (Beyotime Institute of Biotechnology).
Real Time PCR analysis
Total RNA was extracted from transgenic and non-transgenic plants using a guanidine protocol. The extracted RNA was treated with DNase (RNase free) to remove DNA contamination. RNA was quantified by measuring the absorbance at 260 nm, and the quality was checked with agarose gel electrophoresis. Four micrograms of RNA were reverse transcribed with oligo dT primers using M-MLV Reverse Transcriptase (MBI Fermentas) in a volume of 20 ml. Primers were designed for amplification of the RACK1 gene, and the GAPDH gene was used as an internal standard (Table 1). Real-time RT-PCR was repeated three times for each sample. The PCR reaction system (20 μL) consisted of 10.5 μL dd H2O, 0.5 μL Taq, 2 μL buffer, 2 μL 2.5 mmol/L dNTP and 2 μL forward and reverse primers (10 μmol), respectively, and 1 μL template. The cycling parameters were 94ºC for 5 min, followed by 30 cycles of 94ºC for 30 s, 55ºC for 30 s and 72ºC for 30 s, 72ºC for 10 min after 30 cycle. The expressions of RACK1 and GAPDH were quantified according to the amount of their values from the standard curve (Ct).
Table 1.
Primers used in Real-time PCR
| Primer | Name | Sequence |
|---|---|---|
| P1 | RACK1 (forward primer) | 5’-CAAGGTCGGGCAGGAAGAG-3’ |
| P2 | RACK1 (reverse primer) | 5’-TAGAAGGCACAGTCGAGG-3’ |
| P3 | GAPDH (forward primer) | 5’-GTCGGTGTGAACGGATTTG-3’ |
| P4 | GAPDH (reverse primer) | 5’-TCCCATTCTCAGCCTTGAC-3’ |
Western blot analysis
At the end of incubation, the medium was removed, H9C2 cells were harvested and were washed briefly with PBS and immediately lysed in a buffer containing 20 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, and protease and phosphates inhibitors at 4°C for 30 min. The total proteins were quantified using the BCA protein assay kit. Loading buffer was added to cytosolic extracts and boiling for about 5 min, the same amounts of supernatant from each sample were separated by 10% SDS PAGE and transferred to a polyvinylidene difluoride membrane. After blocking with 5% skim milk for 1.5 h in fresh block buffer (TBS-T, 0.1% Tween 20 in Trisbuffered saline) at room temperature, the membrane was probed with anti-RACK1, anti-ENO1, anti-Prx 2, anti-Dsg1, anti-ALDOC, anti-iNOS, anti-CaSR, anti-p-JNK1/2, anti-P-ERK1/2, anti-Cytc, anti-Caspase3, anti-Bcl-2 anti-CaM and anti-CD147 in freshly prepared TBS-T with 3% free-fat milk overnight with gentle agitation at 4°C. The membranes were washed for 3 times with TBS-T and then incubated with a secondary antibody for 1.5 h at room temperature in TBS-T with 3% fat-free milk (horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody, Then, membranes were washed three times with TBS-T for 5 min. The immunoreactive signals were visualized by using an ECL (enhanced chemiluminescence) detection. To quantify the protein expression, the X-ray films were scanned and analysed with ImageJ 1.47i software. The experiment was repeated three times.
Statistical analysis
The experiments were repeated three times in six replicate samples. Data were expressed as mean ± standard deviation. One-way ANOVA followed by Dunnett’s t-test was used for statistical analysis (SPSS 13.0 software, SPSS, USA), and the level of significance was set at P < 0.05 (*), P < 0.01 (**).
Results
Construction of a lentiviral expression vector for RNA interference (RNAi) of RACK1
It was reported that Propranolol protects myocardial cells from I/R injury [12], but the mechanism is still unclear. In our previous studies, RACK1 expression decreased markedly in the present of propranolol especially S-propranolol, so S-propranolol was used in the following studies. In order to investigate the protected mechanisms of propranolol in I/R, RACK1 RNAi plasmids were constructed. Three pairs of human RACK1 gene short hairpin RNA (shRNA) sequences were designed online. After synthesis and annealing, three double-stranded oligonucleotides (dsOligo) were cloned into the pLKO-EGFP plasmid, which were subsequently confirmed by DNA sequencing analysis. The constructs were packed into the recombinant lentivirus Lv-RACK1-shRNA in 293T cells. H9C2 cells were then infected with Lv-RACK1-shRNA. The silencing effect of Lv-RACK1-shRNA in H9C2 cells were validated by real-time PCR and Western-blotting (Figure 1). A significant decrease of RACK1 mRNA (Figure 1A) and protein level (Figure 1B) was observed in H9C2 cells infected with Lv-RACK1-shRNA when compared with that in control cells. The decrease ratios were 67% and 47%, respectively. Our results indicated that an effective Lv-RACK1-shRNA could inhibit the expression of RACK1 in both mRNA and protein levels.
Figure 1.

Construction of a lentiviral expression vector for RNA interference (RNAi) of RACK1. H9C2 cells were cultured as described in Methods. mRNA (A) and protein (B, C) levels of RACK 1 were assessed by real time PCR and western-blot, respectively. GAPDH was used as an internal reference (1: no treatment, 2: infected with control, 3: infected with Lv-RACK1-shRNA). Data were presented as mean ± standard deviation. The experiments were repeated 3 times. Error bars indicate standard deviations. **P < 0.01 compare with 1, ##P < 0.01 compare with 2.
RACK1 RNAi treatment decreased the release of cytokines and chemokines in I/R H9C2 cells
Cytokines and chemokines are low molecular weight polypeptides that allow communication between cells. It is well known that abnormal productions of several cytokines such as tumor necrosis factor (TNF)-α, interleukin-1beta (IL-1b), IL-6 and IL-8 are implicated in the pathogenesis of various inflammatory and autoimmune diseases, including myocardial damage. As shown in Table 2, the levels of TNF-α, IL-1b, IL-6 and IL-8 significantly decreased in I/R H9C2 cells infected with Lv-RACK1-shRNA when compare with I/R H9C2 cell control. The decrease ratios were 31.4%, 31.9%, 36.7% and 34.0%, respectively. After S-propranolol treatment, the levels of TNF-α in I/R H9C2 cells with RACK1 interference were significantly drop to 75.1% and 87.6% of RACK1 interfere group respectively. Similar reductions were observed in the levels of IL-1b, IL-6 and IL-8. Our results suggested that the downregulation of RACK1 decreased the productions of these cytokines in I/R H9C2 cells, which can be augmented by S-propranolol treatment.
Table 2.
TNF-α, IL-1b, IL-6 and IL-8 levels after treatment with S-propranololon or R-propranololon in I/R H9C2 cells
| Item | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| TNF-α | 976.0±13.4 | 714.0±5.1* | 728.8±6.2# | 536.4±6.9## |
| IL-6 | 215.9±1.7 | 143.3±0.9* | 144.9±0.8# | 96.6±0.7## |
| IL-8 | 2617±10 | 1907±9* | 1930±18# | 1325±16## |
| IL-1b | 59.76±0.30 | 40.06±0.32* | 40.45±0.32# | 23.92±0.33## |
P<0.05 compare with 1;
P<0.05 compare with 1;
P<0.01 compare with 2.
1) RACK1 control; 2) Lv-RACK1-shRNA; 3) RACK1 control + S-propranolol; 4) Lv-RACK1-shRNA + S-propranolol.
Effects of RACK 1 RNAi on Ca2+, MMP, ROS, and cell apoptosis
Cardiac myocyte apoptosis is potentially important in many cardiac disorders. To evaluate the effects of RACK1 RNAi on myocardial apoptosis, cell apoptosis were also determined by FACS. MMP is a direct indicator of mitochondrial aerobic respiration efficiency. Intracellular Ca2+ generation is positively correlated with MMP in cells under normal culture conditions, and mitochondrial Ca2+ is another powerful signal for ROS production, which directly results in cell death. The Ca2+, ROS and cell apoptosis were also determined in ischemia/reperfusion H9C2 cell. As shown in Figures 2 and 3, cell apoptosis, Ca2+ and ROS concentration in RACK1 RNAi cells significantly reduced compare to H9C2 cell control after I/R treatment. MMP level was increased in Lv-RACK1-shRNA + S-propranolol group in comparison of H9C2 cell control after I/R treatment. Such effect can be strengthened by the adding of S-propranolol. Our results suggested that RACK1 RNAi and S-propranolol treatment can significantly protect cells from apoptosis.
Figure 2.
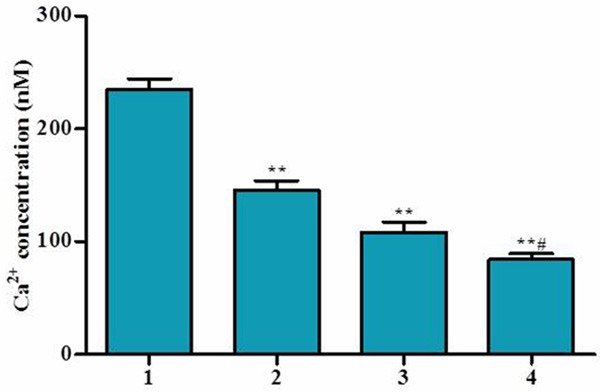
Effect of S-propranolol on Ca2+ in I/R H9C2 cells. Cells were cultured in low glucose DMEM without FBS in a hypoxia chamber, saturated with 5% CO2 for 24 h and following reoxygenation using 10% FBS high glucose DMEM and put in the normal incubating condition for 24 h. Ca2+ concentration was measured. Cells were treated as follow: 1) RACK1 control, 2) Lv-RACK1-shRNA, 3) RACK1 control + S-propranolol, 4) Lv-RACK1-shRNA + S-propranolol. *P < 0.05 versus 1, **P < 0.01 versus 1, #P < 0.05 versus 2, ##P < 0.01 versus 2.
Figure 3.

Effects of S-propranolol on MMP, Ca2+, ROS and cell apoptosis in I/R H9C2 cells were measured by flow cytometers. Cells were cultured in low glucose DMEM without FBS in a hypoxia chamber, saturated with 5% CO2 for 24 h and following reoxygenation using 10% FBS high glucose DMEM and put in the normal incubating condition for 24 h. MMP level (A1, A2), ROS (B1, B2) and cell apoptosis (C1, C2) were measured. Cells were treated as follow: 1) RACK1 control, 2) Lv-RACK1-shRNA, 3) RACK1 control + S-propranolol, 4) Lv-RACK1-shRNA + S-propranolol. *P < 0.05 versus 1, **P < 0.01 versus 1, #P < 0.05 versus 2, ##P < 0.01 versus 2.
Effects of RACK1 RNAi on expressions of CaSR
Calcium-sensing receptor (CaSR) is a G-protein coupled receptor (GPCR) that belongs to family, which can give rise to Ca 2+ release from sarcoplasmic reticulum when it is activated [1]. Content change of Ca2+ in mitochondria is considered as the reason for cell apoptosis [2,3]. In the present study, mRNA expression of CaSR in the RACK1 interfered group was significantly decreased. In addition, in Lv-RACK1-shRNA + S-propranolol group, a marked decrease was observed compared with that of RACK1 control group and Lv-RACK1-shRNA group (Figure 4).
Figure 4.
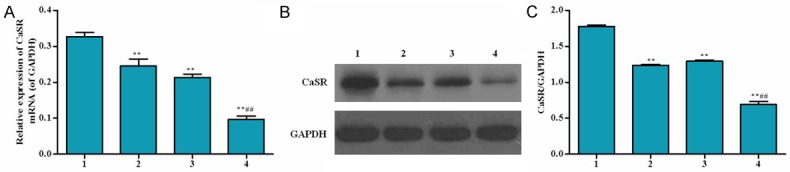
Effect of RACK1 RNAi on mRNA expression and protein level of CaSR in human H9C2 cells. A. mRNA expression of CaSR was detected by real time PCR. B. Protein level of CaSR was explored by western-blot. C. Cells were treated as follow: 1) RACK1 control, 2) Lv-RACK1-shRNA, 3) RACK1 control + S-propranolol, 4) Lv-RACK1-shRNA + S-propranolol. *P < 0.05 versus 1, **P < 0.01 versus 1, #P < 0.05 versus 2, ##P < 0.01 versus 2.
Effects of RACK1 RNAi on cytochrome C release, caspase-3 activity, and JNK activation
I/R can execute apoptosis by activating mitochondrial pathway. The activation of mitochondrial pathway causes the release of cytochrome C from the mitochondria into the cytosol. Cytochrome C in RACK1 interfered group significantly increased in the mitochondria (2.10 fold of control) and markedly reduced to 54% of control in the cytosol. S-propranolol treatment can remarkable improve such changed (Figure 5).
Figure 5.
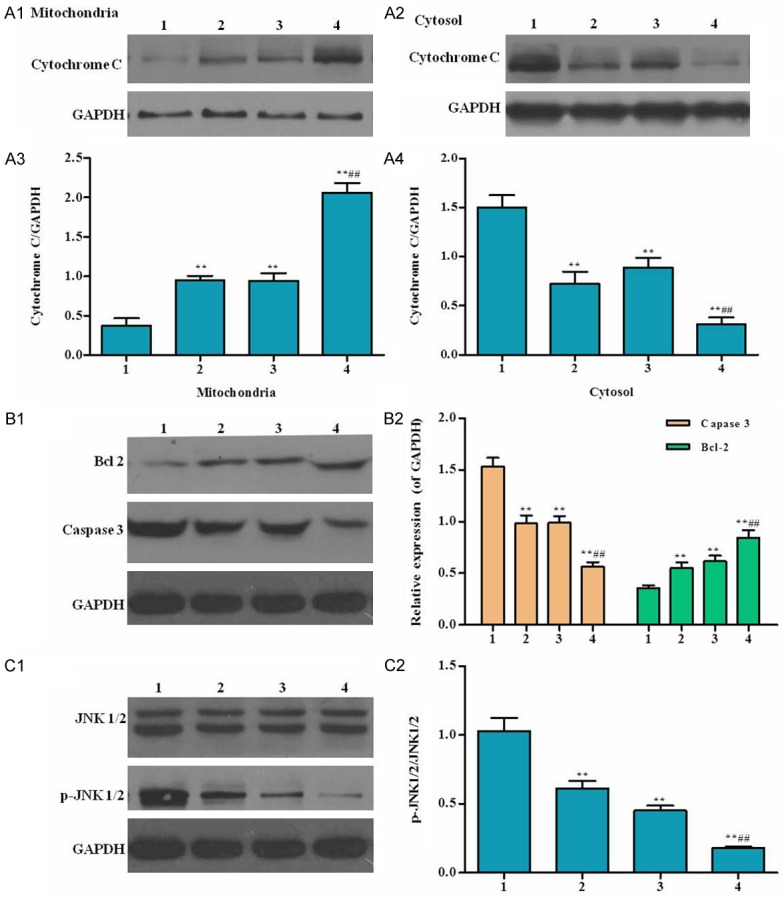
Effects of RACK1 RNAi on cytochrome C release, caspase-3 activity, Bcl 2 activity and JNK activation. Cells were lysed for western blot analysis using antibodies against the indicatedproteins. A1-4. Mitochondrial and cytosolic Cytochrome C; B. Caspase 3 and Bcl 2; C. JNK 1/2 and p-JNK 1/2. Cells were treated as follow: 1) RACK1 control, 2) Lv-RACK1-shRNA, 3) RACK1 control + S-propranolol, 4) Lv-RACK1-shRNA + S-propranolol. *P < 0.05 versus 1, **P < 0.01 versus 1, #P < 0.05 versus 2, ##P < 0.01 versus 2.
Caspase 3 was considered as an effector of apoptosis (Kumar, 1997). Caspase-3 activated by cytochrome C through the interaction with Apaf-1 and caspase-9, resulting in apoptosis. Bcl-2 is another key regulator of apoptosis. In our study (Figure 5B1, 5B2), Caspase 3 significantly decreased and Bcl-2 is increased in RACK1 interfered group. Yue et al. (39) found that JNK mediate apoptosis in cardiomyocytes subjected to I/R. In our study (Figure 5C1, 5C2), significantly decrease of phosphorylation of JNK was observed in RACK1 RNAi group compared to control. Total JNK protein expression in RACK1 interfered group are similar with I/R control. S-propranolol treatment can augment the reduction of JNK phosphorylation.
Discussion and conclusion
RNAi is a widely used method to study gene functions. In the present study, RACK1 mRNA and protein expression was decreased in the interference model group. The interference efficiency was above 47% (Figure 1) and could be used in our following studies.
Many in vivo studies have demonstrated that TNF-α, IL-1b, IL-6, and IL-8 are all important components of the pro-inflammatory response and intraocular inflammation [15]. Oxidative stress is associated with many systemic inflammatory diseases, and free-radical production leads to the formation of self-propagating lipid peroxidation [16]. The pro-inflammatory cytokines interleukin (IL)-lb and IL-6 have been shown to be up-regulated in the myocardium early after injury, tumor necrosis factor (TNF-α), IL-lb and IL-6 are induced in the myocardium after treatment with norepinephrine (NE) by a receptor mediated mechanism [17]. In the present sduty, the levels of TNF-α, IL-1b, IL-6 and IL-8 significantly decreased separately and in combination. Our data suggested that s-propranolol may be involved in the increase of cytokine expression and remodeling after myocardial infarction.
ROS play a key role in promoting cytochromecrelease from the mitochondria, and cytochromecin the cytoplasm triggers activation of the caspase cascade that ultimately leads to apoptosis. Cytochrome C release from mitochondria proceeds by a two-step process: dissociation of cytochrome c from cardiolipin in the inner mitochondrial membrane (IMM), followed by release of cytochrome c through the outer mitochondrial membrane (OMM). Impairment of mitochondria leading to collapse of mitochondrial membrane potential causes release of cytochrome C and activates caspase-9 leading to activation of caspase-3. Then intranucleosomal DNA fragmentation is finally induced by endonucleases and the final steps of apoptosis occur. It leads to opening of mitochondrial membrane permeability transition pore and the release of proapoptotic proteins including cytochrome C from the mitochondrial matrix. The release is known to be caused by mitochondrial dysfunction because the membrane potential could not be properly regulated in such a case. Cytochrome C activates caspase-3 through the interaction with Apaf-1 and caspase-9, resulting in apoptosis.
Calcium is taken up into mitochondria via a uniporter (UP) in the IMM, and UP has been identified as a Ca2+ selective ion channel and elevation of mitochondrial Ca2+ and ROS production is thought to play an important part in cell death associated with ischemia-reperfusion. Induction of cell death as a consequence of mitochondrial Ca2+ uptake, and Ca2+ is effectively modulated by several factors, including ROS, pH, MMP (P. Bernardi, 1999).
Myocardial mitogen-activated protein kinases (MAPKs) can be activated by I/R, and they may play important roles in the evolution of ischemic injury Yue et al. [18]. examined the interaction among the MAPKs during I/R in neonatal cardiomyocytes and found that ERK, p38, and JNK pathways were activated rapidly and transiently by ischemia. The JNK pathway is more important than the p38 pathway in signaling oxidative stress-induced cell injury, using a cardiac muscle cell line. In the present study, RACK1 RNAi and S-propranolol, separately and in combination significantly decreased the phosphorylation of JNK in I/R H9C2 cells (Figure 4). These data indicated that RACK1 RNAi and S-propranolol protected cells from apoptosis via the inhibition of JNK activation.
Acknowledgements
This study was supported by the General Program of Science and Technology Project of Yunnan Province, China (2011FB216, 2011FB217), and the Scientific Research Project of research institutions of medical and health unit in Yunnan Province (2014NS247, 2014NS246).
Disclosure of conflict of interest
None.
References
- 1.Armiger L, Gavin J. Changes in the microvasculature of ischemic and infarcted myocardium. Lab Invest. 1975;33:51–56. [PubMed] [Google Scholar]
- 2.Ockaili R, Natarajan R, Salloum F, Fisher BJ, Jones D, Kukreja RC. HIF-1 activation attenuates postischemic myocardial injury: role for heme oxygenase-1 in modulating microvascular chemokine generation. Am J Physiol Heart Circ Physiol. 2005;289:H542–H548. doi: 10.1152/ajpheart.00089.2005. [DOI] [PubMed] [Google Scholar]
- 3.Mccord J. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 4.Szendrei L, Turoczi T, Kovacs P, Vecsernyes M, Das DK, Tosaki A. Mitochondrial gene expression and ventricular fibrillation in ischemic/reperfused nondiabetic and diabetic myocardium. Biochem Pharmacol. 2002;63:543–552. doi: 10.1016/s0006-2952(01)00913-3. [DOI] [PubMed] [Google Scholar]
- 5.Das S, Powell SR, Wang P, Divald A, Nesaretnam K, Tosaki A, Cordis GA, Maulik N, Das DK. Cardioprotection with palm tocotrienol: antioxidant activity of tocotrienol is linked with its ability to stabilize proteasomes. Am J Physiol Heart Circ Physiol. 2005;289:H361–H367. doi: 10.1152/ajpheart.01285.2004. [DOI] [PubMed] [Google Scholar]
- 6.Liu HR, Gao E, Hu A, Tao L, Qu Y, Most P, Koch WJ, Christopher TA, Lopez BL, Alnemri ES. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111:90–96. doi: 10.1161/01.CIR.0000151613.90994.17. [DOI] [PubMed] [Google Scholar]
- 7.Kingma JG Jr, Denniss AR, Hearse DJ, Downey JM, Yellon DM. Limitation of infarct size for 24 hours by combined treatment with allopurinol plus verapamil during acute myocardial infarction in the dog. Circulation. 1987;75:V25–33. [PubMed] [Google Scholar]
- 8.Black JW, Crowther A, Shanks R, Smith L, Dornhorst A. A new adrenergic: beta-receptor antagonist. Lancet. 1964;283:1080–1081. doi: 10.1016/s0140-6736(64)91275-9. [DOI] [PubMed] [Google Scholar]
- 9.Rabkin R, Stables DP, Levin NW, Suzman MM. The prophylactic value of propranolol in angina pectoris. Am J Cardiol. 1966;18:370–380. doi: 10.1016/0002-9149(66)90056-7. [DOI] [PubMed] [Google Scholar]
- 10.Wolfson S, Gorlin R. Cardiovascular Pharmacology of Propranolol in Man. Circulation. 1969;40:501–511. doi: 10.1161/01.cir.40.4.501. [DOI] [PubMed] [Google Scholar]
- 11.Maroko PR, Braunwald E. Effects of metabolic and pharmacologic interventions on myocardial infarct size following coronary occlusion*. Acta Med Scand Suppl. 1976;199:125–136. doi: 10.1111/j.0954-6820.1976.tb05874.x. [DOI] [PubMed] [Google Scholar]
- 12.Kloner RA, Fishbein MC, Cotran RS, Braunwald E, Maroko PR. The effect of propranolol on microvascular injury in acute myocardial ischemia. Circulation. 1977;55:872–880. doi: 10.1161/01.cir.55.6.872. [DOI] [PubMed] [Google Scholar]
- 13.Lupo J, Conti A, Sueur C, Coly PA, Couté Y, Hunziker W, Burmeister WP, Germi R, Manet E, Gruffat H. Identification of new interacting partners of the shuttling protein ubinuclein (Ubn-1) Exp Cell Res. 2012;318:509–520. doi: 10.1016/j.yexcr.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Fire A. RNA-triggered gene silencing. Trends in Genetics. 1999;15:358–363. doi: 10.1016/s0168-9525(99)01818-1. [DOI] [PubMed] [Google Scholar]
- 15.Hoekzema R, Murray P, Kijlstra A. Cytokines and intraocular inflammation. Curr Eye Res. 1990;9:207–211. doi: 10.3109/02713689008999443. [DOI] [PubMed] [Google Scholar]
- 16.Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- 17.Deten A, Volz HC, Holzl A, Briest W, Zimmer HG. Biochemistry of Hypertrophy and Heart Failure. Springer; 2003. Effect of propranolol on cardiac cytokine expression after myocardial infarction in rats; pp. 127–137. [PubMed] [Google Scholar]
- 18.Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2014;97:276–285. doi: 10.1007/s00395-002-0364-9. [DOI] [PubMed] [Google Scholar]