

Abstract
Albumin-bound fatty acids is the main cause of renal damage, PPARα is responsible in the metabolism of fatty acids. Previous study found that PPARα played a protective role in fatty acids overload associated tubular injury. The aim of the present study is to investigate whether fenofibrate, a PPARα ligands, could contribute to the renoprotective action in fatty acids overload proximal tubule epithelial cells. We observed in HK-2 cells that fenofibrate significantly inhibited fatty acids bound albumin (FA-BSA) induced up-regulation of MCP-1 and IL-8. Treatment with fenofibrate attenuated renal oxidative stress induced by FA-BSA as evidenced by decreased MDA level, increased SOD activity and catalase, GPx-1 expression. FA-BSA induced apoptosis of HK-2 cells were also obviously prevented by fenofibrate. Furthermore, fenofibrate significantly increased the expression of PPARα mRNA and protein in FA-BSA treated cells. Finally, the activation of NF-kB induced by FA-BSA was markedly suppressed by fenofibrate. Taken together, our study describes a renoprotective role of fenofibrate in fatty acids associated tubular toxicity, and the transcriptional activation of PPARα and suppression of NF-kB were at least partially involved.
Keywords: Chronic kidney disease, fatty acids, peroxisome proliferator-activated receptor α, fenofibrate, proximal tubule epithelial cells, apoptosis
Introduction
Chronic kidney disease (CKD) is a major disease characterized by heterogeneous disorders in kidney that seriously threatened to human health. Approximately 13% of the world’s population are suffering from CKD [1]. In china, CKD patients accounts for about 10.25% of the total population, and the incidence is rising year by year [2]. CKD is considered to be a typically progressive disease, and it is probably irreversible when developed to end-stage renal disease (ESRD) [3]. At present, dialysis and transplantation are main therapies of ESRD, but the high cost severely restricted their applicability. Therefore, there is an urgent need to develop novel and effective therapies to prevent the progression of CKD.
Recently, free fatty acids that binding to albumin was reported to be the main cause of renal damage, rather than albumin itself [4,5]. Peroxisome proliferator-activated receptor α (PPARα), a member of the nuclear hormone receptor super family, is intensively expressed in the kidney and closely related to fatty acid oxidation by the regulation of several lipolysis enzymes [6,7]. PPARα deficiency results in fatty acid metabolism disorder as well as proximal tubular dysfunction [8,9], therefore, PPARα may play a protective role in fatty acid associated tubular toxicity.
Fenofibrate is a specific ligands of PPARα that could specifically bind to and activate the transcription of PPARα [10]. It is widely used for the treatment of hyperlipidemia for its lipid metabolism promotion properties [11]. Animal studies have demonstrated that fenofibrate attenuated renal lipotoxicity and protected renal function by increasing fatty acid oxidation as well as preventing cell death in diabetic nephropathy mice and hypertensive rats [12,13]. Thus, we hypothesized that fenofibrate could activate PPARα and protect kidney from fatty acids induced damage. To test our hypothesis, an fatty acids-bound albumin overload model of human proximal tubule epithelial cells was employed to examine the effect of fenofibrate in inflammatory reactions, oxidative stress and apoptosis.
Materials and methods
Cell culture
Immortalized human proximal tubule epithelial cell line HK-2 was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA) at 37°C in a humidified atmosphere of 5% CO2.
MTT assay
To determine the optimal concentration of fatty acids bounded BSA (FA-BSA), HK-2 cells were seeded into 96-well plates (2×103 cells/well) and grown for 24 h in a 37°C, 5% CO2 incubator. FA-BSA (Solarbio, Beijing, China) was added at a final concentration of 0 g/l, 1.25 g/l, 2.5 g/l, 5 g/l, 10 g/l or 20 g/l. Then, 0.2 g/l MTT working solution (Sigma-Aldrich Int, Louis, MO, USA) was added at different time intervals. 4 h latter, the crystals was dissolved with 200 μl dimethyl sulfoxide (DMSO), and the value of OD490 was measured by a microplate reader (ELX-800; Bio-TEK Instruments Inc., Winooski, VT, USA). After that, cells were pretreated with 0 µmol/l, 25 µmol/l, 50 µmol/l, 100 µmol/l or 200 µmol/l fenofibrate (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h, then, the optimal concentration of FA-BSA was added, after co-incubation for 24 h, the employed concentration of fenofibrate was determined by MTT assay as mentioned above.
Cell treatment
HK-2 cells were plated in 6-well plates and cultured at 37°C, 5% CO2 for 24 h. The experiment was set as follows: control group, HK-2 cells without any treatment; FA-BSA group, HK-2 cells incubated with 10 g/l FA-BSA; Fen group, HK-2 cells incubated with 100 µmol/l fenofibrate; FA-BSA + Fen group, HK-2 cells pretreated with 100 µmol/l fenofibrate for 2 h followed by 10 g/l FA-BSA incubation. 24 h later, cells from each group were centrifuged at 1000 rpm for 10 min, the cell pellet and supernatant were collected respectively for further analysis.
Measurement of IL-8, MCP-1, MDA and SOD
The levels of IL-8 and MCP-1 in supernatant were measured by ELISA kits (Multi Sciences, Hangzhou, China) according to manufacturer’s instructions. Cell pellets from each group were resuspended by 600 µl phosphate buffer saline (PBS, Doublehelix Biology Science, Wuhan, China) and freezing and thawing in liquid nitrogen for three times. Cell lysates was centrifuged at 12000 rpm for 10 min and the supernatant was collected. Methane dicarboxylic aldehyde (MDA) concentration and superoxide dismutase (SOD) activity was determined by commercial kits (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) as per the instructions.
Detection of cell apoptosis
Cells from each group were subjected to apoptosis detection by using a Annexin V-FITC Apoptosis Detection Kit (Key GEN, Nanjing, China). In brief, cells were resuspended with Annexin V-FITC binding buffer and incubated with Annexin V-FITC and propidium iodide at room temperature for 15 min in dark. Then, the apoptosis cells were analyzed by flow cytometry (BD Accuri C6, BD Biosciences, Ann Arbor, MI, USA).
Hoechst 33258 staining
HK-2 cells were seeded on 12-well plates with coverslip and subjected to different treatment. After fixing, slides of cells were incubated with Hoechst 33258 staining solution (Beyotime) at room temperature for 5 min. Then the slides were washed two times by PBS. Finally, the stained cells were observed under a fluorescence microscope (BX53, Olympus, Tokyo, Japan).
Western blot analysis
Cells from each group were lysed by RIPA buffer (Beyotime, Haimen, China) containing 1% PMSF, total proteins were collected by centrifugation. Cytoplasm and nucleus proteins were isolated using a nuclear extraction kit (Beyotime). The protein concentration was determined by BCA method (Beyotime). Equal amounts of proteins were separated using 10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). After blocking, the membranes were then hybridized with primary antibodies (catalase, 1:200 diluted; GPx-1, 1:200 diluted; PPARα, 1:200 diluted, purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA. bax, 1:400 diluted; bcl-2, 1:400 diluted; NF-kB, 1:400 diluted, purchased from Boster, Wuhan, China; p-IkB, 1:500 diluted, purchased from Bioss, Beijing, China), followed by incubation with HRP conjugated secondary antibodies. The specific protein bands were detected by using an enhanced chemiluminescent kit (Qihai Biotec, Shanghai, China).
Real-time PCR
Total RNA of HK-2 cells were extracted using RNA sim-ple Total RNA Kit (Bioteke Corporation, Beijing, China) and reverse-transcribed by Super M-MLV reverse transcriptase (Bioteke Corporation, Beijing, China). SYBER Green based Real-time PCR was performed using 2× Power Taq PCR MasterMix (Bioteke Corporation). Specific primers for PPARα and β-actin were synthesized as follows: PPARα F: 5’-TCATCACGGACACGCTTTC-3’, R: 5’-CCCGCAGATTCTACATTCG-3’; β-actin F: 5’-CTTAGTTGCGTTACACCCTTTCTTG-3’, R: 5’-CTGTCACCTTCACCGTTCCAGTTT-3’. The mRNA level of PPARα was analyzed by 2-ΔΔCt method, β-actin was employed as an internal control.
Statistical analysis
Results were presented as mean ± standard deviation. Differences between groups were analyzed by one way analysis of variance (ANOVA) and the Bonferroni post hoc test. Data analysis was performed using a microcomputer-assisted program with SPSS 16.0 statistical analysis software (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to be statistically significant.
Results
Fenofibrate attenuates FA-BSA induced growth inhibition of HK-2 cells
HK-2 cells were treated with different concentrations of FA-BSA, the employed concentration was detected by MTT assay. As shown in Figure 1A, FA-BSA inhibited HK-2 cell viability in a dose-and time-dependent manner. Treatment with 10 g/l FA-BSA resulted in a significant inhibition of cell proliferation. Thus, 10 g/l was employed as an experiment concentration of FA-BSA. The effective concentration of fenofibrate was also determined by MTT assay (Figure 1B), 50 μmol/l fenofibrate significantly reversed the inhibition effect of FA-BSA on cell viability, and treatment with 100 μmol/l fenofibrate reached the inflexion, thus 100 μmol/l was chosen as an experiment concentration of fenofibrate.
Figure 1.
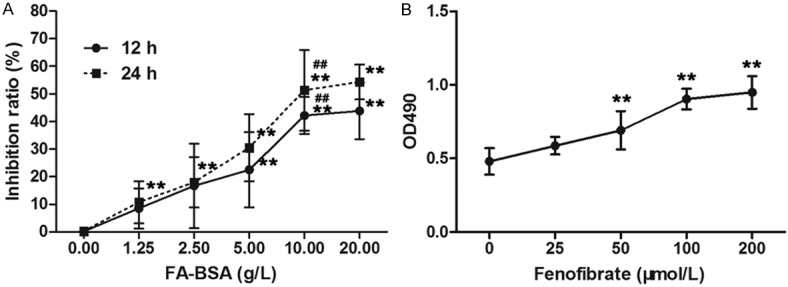
Fenofibrate prevented HK-2 cells from FA-BSA induced growth inhibition. The inhibition ratio of HK-2 cells (A) and cell viability (B) were detected by MTT assay. Data were expressed as mean ± standard deviation. **P<0.01 vs. untreated group, ##P<0.01 vs. 5 g/l group.
Fenofibrate ameliorates FA-BSA induced inflammatory reaction and oxidative stress in HK-2 cells
To investigate the effect of fenofibrate on FA-BSA induced inflammatory, the levels of MCP-1 and IL-8 were determined. As expected, the over-expression of MCP-1 and IL-8 in HK-2 cells induced by FA-BSA was dramatically decreased by fenofibrate treatment (Figure 2A, 2B). Moreover, FA-BSA induced oxidative damage was also restored by fenofibrate treatment that characterized by decreased level of lipid peroxidation products, MDA, and enhanced activity of SOD (Figure 2C, 2D). Actually, the expressions of major antioxidant enzymes, catalase and GPx-1 that strongly reduced by FA-BSA stimulation were also evidently up-regulated by fenofibrate treatment. Thus, these data suggest that fenofibrate could effectively inhibit FA-BSA induced inflammatory reactions and oxidative stress in HK-2 cells.
Figure 2.
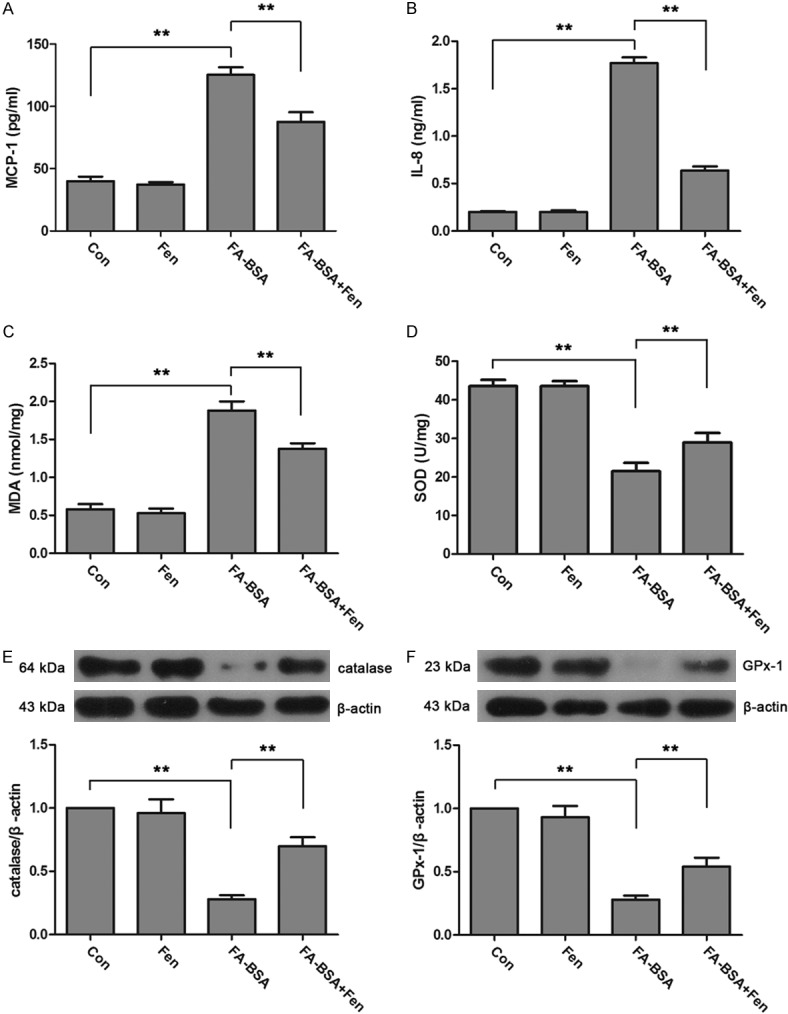
Fenofibrate inhibited FA-BSA induced inflammation and oxidative stress in HK-2 cells. Concentrations of MCP-1 (A), IL-8 (B), MDA (C) and activity of SOD (D) were measured by using commercial kits. Expressions of catalase (E) and GPx-1 (F) were examined by western blot. Data were expressed as mean ± standard deviation. P<0.01 is represented by double asterisks.
Fenofibrate inhibits FA-BSA induced apoptosis of HK-2 cells
To test whether fenofibrate exerts protective effects in FA-BSA induced apoptosis, we further evaluated the apoptosis status of HK-2 cells. Flow cytometry showed that the proportion of apoptotic cells was significantly up-regulated by FA-BSA treatment, whereas it was markedly reduced by fenofibrate (Figure 3A). These changes were further confirmed by Hoechst 33258 staining. As shown in Figure 3B, there are more apoptosis cells in FA-BSA group that identified by condensation of nuclear chromatins, however, it was obviously reduced by fenofibrate treatment. Furthermore, the levels of apoptosis related proteins, bcl-2 and bax, were measured by western blot (Figure 3C, 3D). The data showed that treatment with FA-BSA resulted in a significant up-regulation of bax and down-regulation of bcl-2, and these changes were markedly reversed by fenofibrate. In summary, fenofibrate is effective in the inhibition of FA-BSA induced renal proximal tubular cells apoptosis.
Figure 3.
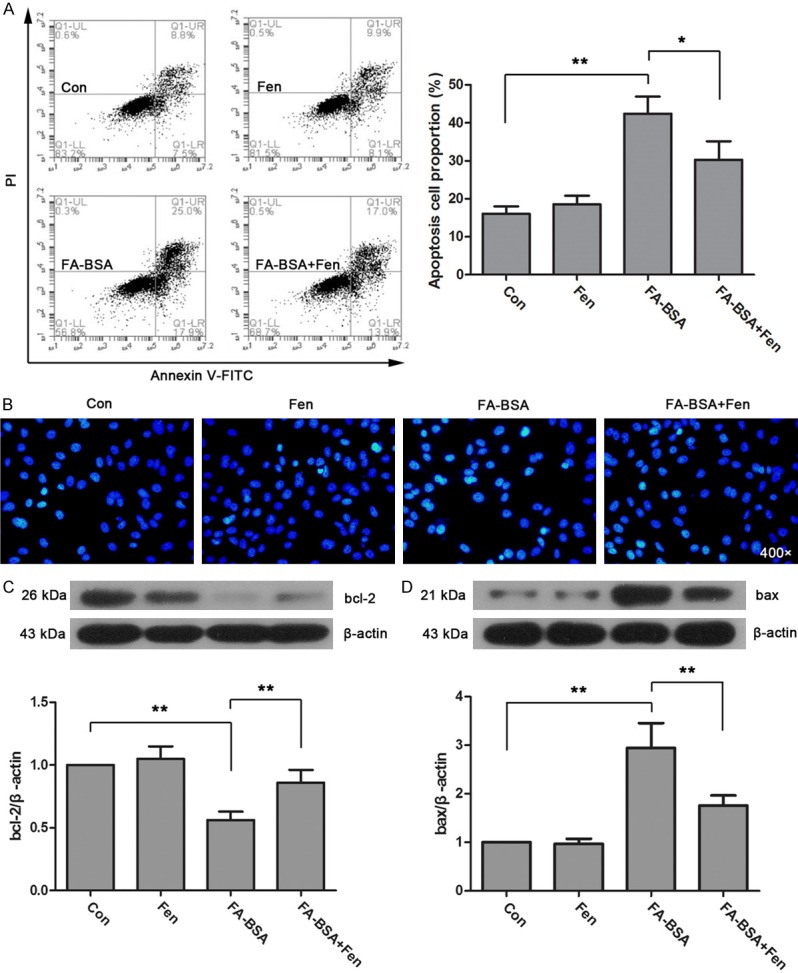
Fenofibrate protected HK-2 cells from FA-BSA induced apoptosis. Apoptosis of HK-2 cells were detected by flow cytometry (A) and Hoechst 33258 staining (B). The expression of bcl-2 (C) and bax (D) was determined by western blot. Data were expressed as mean ± standard deviation. P<0.05 is represented by single asterisks. P<0.01 is represented by double asterisks.
Fenofibrate activates PPARα and suppresses NF-kB pathway in HK-2 cells
To determine whether PPARα is involved in the protective action of fenofibrate in FA-BSA induced HK-2 cell damage, the expression of PPARα was measured by real-time PCR and western blot (Figure 4A, 4B). FA-BSA overload caused a marked decrease of PPARα mRNA and protein, but it was significantly up-regulated by fenofibrate treatment. It is reported that renal NF-kB signaling pathway is regulated by PPARα [14], thus we examined the activity of NF-kB to investigate the mechanism of how fenofibrate protected HK-2 cells from FA-BSA induced injury. As shown in Figure 4C and 4D, the up-regulated level of cytoplasmic p-IkB and nuclear NF-kB induced by FA-BSA in HK-2 cells was dramatically reduced by fenofibrate treatment. These results indicated that PPARα and NF-kB pathway may be a pivotal mediator in fenofibrate-mediated protection of FA-BSA induced HK-2 injury.
Figure 4.
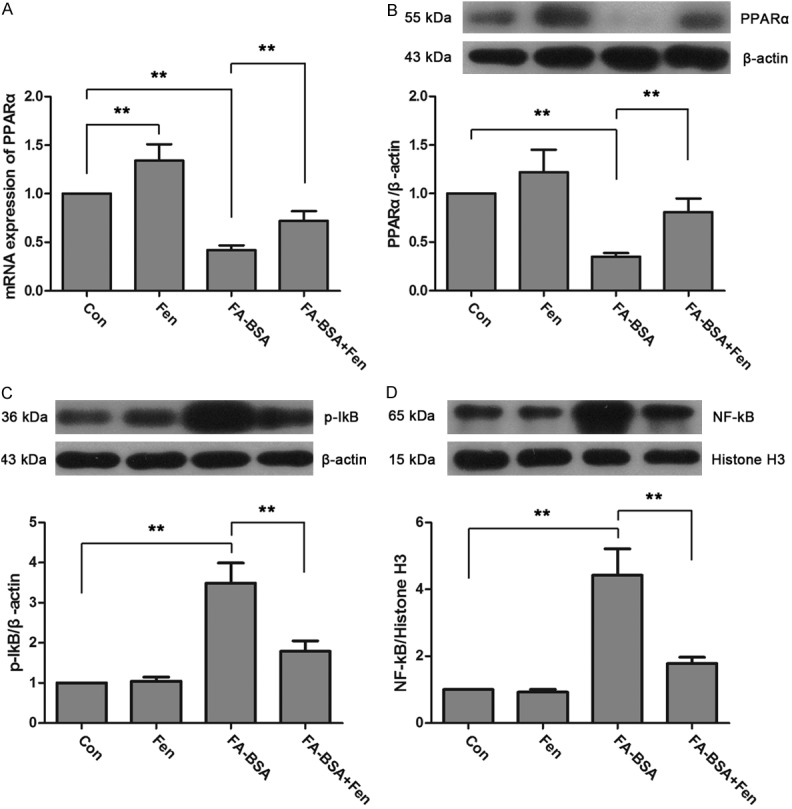
Fenofibrate activated of PPARα transcription and suppressed NF-kB activation. The expressions of PPARα mRNA (A) and protein (B) were detected by real-time PCR and western blot. The expressions of cytoplasmic p-IkB (C) and nuclear NF-kB (D) were determined by western blot. Data were expressed as mean ± standard deviation. P<0.01 is represented by double asterisks.
Discussion
The present study found that fenofibrate reduced the over-expression of inflammatory mediators and inhibited oxidative stress that induced by free fatty acids overload in HK-2 cells. Meanwhile, FA-BSA induced apoptosis of HK-2 cells was also inhibited by fenofibrated treatment. These protective effects of fenofibrated appeared to be associated with up-regulated PPARα expression and suppressed NF-kB activation.
Previous study has profiled in proximal tubular cells that fatty acids bound albumin aggravates oxidative stress and inflammatory responses much more intensively than albumin alone [15]. Animal experiment also showed that intraperitoneally injected with fatty acids binding albumin resulted in severe tubulointerstitial damage [4]. In the present study, HK-2 cells treated with FA-BSA exhibited obviously growth inhibition in a dose-and time-dependent manner, these data further confirmed that albumin binding free fatty acids is responsible in renal damage.
Inflammation and oxidant stress are closely related to the conversion of moderate to severe CKD [16]. MCP-1 and IL-8 are key proinflammatory chemokines that involved in the recruitment and activation of neutrophils, T cells and monocytes [17]. MDA is a index of lipid peroxidation [18], SOD, catalase and GPx-1 are several constitutive amounts of antioxidant enzymes [9]. We found in FA-BSA treated HK-2 cells that fenofibrate reduced MCP-1, IL-8 and MDA level, enhanced SOD activity, and up-regulated catalase and GPx-1 expression, indicating that fenofibrate could attenuate FA-BSA induced inflammation and oxidative stress. Similar conclusions were also obtained in spontaneously hypertensive rats, that fenofibrate protects hypertensive associated kidney injury by the inhibition of oxidative stress [19].
Apoptosis could be observed in the pathogenesis of a variety of kidney diseases, such as diabetic nephropathy [20], hypertensive nephrosclerosis [21] and obestity-related renal disease [22]. It is reported that fatty acids is a chief instigators of renal apoptosis both in vivo and in vitro [23-25]. In the present study, apoptosis was significantly increased in FA-BSA treated group, and it was dramatically reduced by fenofibrate. These results are in line with previous study showing that fenofibrate plays a protective role in high glucose, oxidative stress or high-fat diet induced cell apoptosis [26-28]. The anti-apoptotic protein bcl-2 and pro-apoptotic protein bax are main members of bcl-2 family, that critical in the regulation of apoptosis [29]. In this study, the expression of bcl-2 was inhibited and the level of bax was increased by FA-BSA treatment, however, the changes of bcl-2 and bax were significantly reversed by fenofibrate, these observations explained reasonably on how fenofibrate protected HK-2 cells from FA-BSA induced apoptosis.
The main function of PPARα in proximal tubules is to regulate the metabolism of fatty acids [30]. PPARα-null mice injecting with fatty acids bound albumin exhibited greater tubular injury than wild type mice suggesting a protective role of PPARα in fatty acids associated tubular toxicity [9]. Recently, Takahashi et al. [31] described a renoprotective effect of fibrates, also a PPARα agonist, in fatty acid-induced tubule toxicity by the counteraction of PPARα deterioration. We found in the present study that, FA-BSA over-load resulted in a dramatic reduced of PPARα expression, whereas, it was significantly restored by fenofibrate treatment. Thus, fenofibrate may protect renal from fatty acid induced injury by the activation of PPARα transcription. Further more, a in vitro study found that PPARα mitigated cardiomyocyte hypertrophy by the inhibition of NF-kB activation [32]. In our study, the activated NF-kB signal induced by FA-BSA overload was significantly suppressed by fenofibrate, these results were partly consistent with previous study showing that fenofibrate reduced tubulointerstitial fibrosis and inflammation by the inhibition of NF-kB [33].
In conclusion, our study described a protective role of fenofibrate in fatty acid bound albumin induced inflammation, oxidative stress and apoptosis of human proximal tubule epithelial cells. Moreover, the renoprotective effect of fenofibrate is associated with the activation of PPARα transcription and NF-kB suppression. These results extended our previous observations and suggest a novel therapeutic strategy for preventing the progression of CKD.
Acknowledgements
This work has been supported by two grants: National Natural Science Foundation of China (Project number: 81200656) and Doctoral Scientific Research Foundation of Liao Ning Province (Project number: 20111097).
Disclosure of conflict of interest
None.
References
- 1.Benigni A, Morigi M, Remuzzi G. Kidney regeneration. Lancet. 2010;375:1310–1317. doi: 10.1016/S0140-6736(10)60237-1. [DOI] [PubMed] [Google Scholar]
- 2.Xu R, Zhang L, Zhang P, Wang F, Zuo L, Wang H. Comparison of the prevalence of chronic kidney disease among different ethnicities: Beijing CKD survey and American NHANES. Nephrol Dial Transplant. 2009;24:1220–1226. doi: 10.1093/ndt/gfn609. [DOI] [PubMed] [Google Scholar]
- 3.Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379:165–180. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 4.Kamijo A, Kimura K, Sugaya T, Yamanouchi M, Hase H, Kaneko T, Hirata Y, Goto A, Fujita T, Omata M. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002;62:1628–1637. doi: 10.1046/j.1523-1755.2002.00618.x. [DOI] [PubMed] [Google Scholar]
- 5.Ruggiero C, Elks CM, Kruger C, Cleland E, Addison K, Noland RC, Stadler K. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am J Physiol Renal Physiol. 2014;306:F896–906. doi: 10.1152/ajprenal.00484.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293–22299. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 8.Kamijo Y, Hora K, Tanaka N, Usuda N, Kiyosawa K, Nakajima T, Gonzalez FJ, Aoyama T. Identification of functions of peroxisome proliferator-activated receptor alpha in proximal tubules. J Am Soc Nephrol. 2002;13:1691–1702. doi: 10.1097/01.asn.0000018403.61042.56. [DOI] [PubMed] [Google Scholar]
- 9.Kamijo Y, Hora K, Kono K, Takahashi K, Higuchi M, Ehara T, Kiyosawa K, Shigematsu H, Gonzalez FJ, Aoyama T. PPARalpha protects proximal tubular cells from acute fatty acid toxicity. J Am Soc Nephrol. 2007;18:3089–3100. doi: 10.1681/ASN.2007020238. [DOI] [PubMed] [Google Scholar]
- 10.Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–2093. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 11.Sahebkar A, Watts GF. Challenges in the treatment of hypertriglyceridemia: glass half empty or half full? Expert Rev Clin Pharmacol. 2015;8:363–366. doi: 10.1586/17512433.2015.1045880. [DOI] [PubMed] [Google Scholar]
- 12.Park CW, Zhang Y, Zhang X, Wu J, Chen L, Cha DR, Su D, Hwang MT, Fan X, Davis L, Striker G, Zheng F, Breyer M, Guan Y. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–1517. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 13.Shin SJ, Lim JH, Chung S, Youn DY, Chung HW, Kim HW, Lee JH, Chang YS, Park CW. Peroxisome proliferator-activated receptor-alpha activator fenofibrate prevents high-fat diet-induced renal lipotoxicity in spontaneously hypertensive rats. Hypertens Res. 2009;32:835–845. doi: 10.1038/hr.2009.107. [DOI] [PubMed] [Google Scholar]
- 14.Kono K, Kamijo Y, Hora K, Takahashi K, Higuchi M, Kiyosawa K, Shigematsu H, Gonzalez FJ, Aoyama T. PPARα attenuates the proinflammatory response in activated mesangial cells. Am J Physiol Renal Physiol. 2009;296:F328–F336. doi: 10.1152/ajprenal.00484.2007. [DOI] [PubMed] [Google Scholar]
- 15.Ishola DA Jr, Post JA, van Timmeren MM, Bakker SJ, Goldschmeding R, Koomans HA, Braam B, Joles JA. Albumin-bound fatty acids induce mitochondrial oxidant stress and impair antioxidant responses in proximal tubular cells. Kidney Int. 2006;70:724–731. doi: 10.1038/sj.ki.5001629. [DOI] [PubMed] [Google Scholar]
- 16.Oberg BP, McMenamin E, Lucas FL, McMonagle E, Morrow J, Ikizler TA, Himmelfarb J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004;65:1009–1016. doi: 10.1111/j.1523-1755.2004.00465.x. [DOI] [PubMed] [Google Scholar]
- 17.Shah SV, Baliga R, Rajapurkar M, Fonseca VA. Oxidants in chronic kidney disease. J Am Soc Nephrol. 2007;18:16–28. doi: 10.1681/ASN.2006050500. [DOI] [PubMed] [Google Scholar]
- 18.Agarwal R. Chronic kidney disease is associated with oxidative stress independent of hypertension. Clin Nephrol. 2004;61:377–383. doi: 10.5414/cnp61377. [DOI] [PubMed] [Google Scholar]
- 19.Hou X, Shen YH, Li C, Wang F, Zhang C, Bu P, Zhang Y. PPARalpha agonist fenofibrate protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress and MAPK activity. Biochem Biophys Res Commun. 2010;394:653–659. doi: 10.1016/j.bbrc.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 20.Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, Giannoni M, Rimoldi L, Lauria F, Miji M, Deferrari G, Garibotto G. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007;72:1262–1272. doi: 10.1038/sj.ki.5002531. [DOI] [PubMed] [Google Scholar]
- 21.Ying WZ, Sanders PW. Cytochrome c mediates apoptosis in hypertensive nephrosclerosis in Dahl/Rapp rats. Kidney Int. 2001;59:662–672. doi: 10.1046/j.1523-1755.2001.059002662.x. [DOI] [PubMed] [Google Scholar]
- 22.Hunley TE, Ma LJ, Kon V. Scope and mechanisms of obesity-related renal disease. Curr Opin Nephrol Hypertens. 2010;19:227–234. doi: 10.1097/MNH.0b013e3283374c09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas ME, Harris KP, Walls J, Furness PN, Brunskill NJ. Fatty acids exacerbate tubulointerstitial injury in protein-overload proteinuria. Am J Physiol Renal Physiol. 2002;283:F640–647. doi: 10.1152/ajprenal.00001.2002. [DOI] [PubMed] [Google Scholar]
- 24.Urahama Y, Ohsaki Y, Fujita Y, Maruyama S, Yuzawa Y, Matsuo S, Fujimoto T. Lipid droplet-associated proteins protect renal tubular cells from fatty acid-induced apoptosis. Am J Pathol. 2008;173:1286–1294. doi: 10.2353/ajpath.2008.080137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arici M, Chana R, Lewington A, Brown J, Brunskill NJ. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-gamma. J Am Soc Nephrol. 2003;14:17–27. doi: 10.1097/01.asn.0000042167.66685.ea. [DOI] [PubMed] [Google Scholar]
- 26.Chung HW, Lim JH, Kim MY, Shin SJ, Chung S, Choi BS, Kim HW, Kim YS, Park CW, Chang YS. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARalpha-FoxO3a-PGC-1alpha pathway. Nephrol Dial Transplant. 2012;27:2213–2225. doi: 10.1093/ndt/gfr613. [DOI] [PubMed] [Google Scholar]
- 27.Zanetti M, Stocca A, Dapas B, Farra R, Uxa L, Bosutti A, Barazzoni R, Bossi F, Giansante C, Tedesco F, Cattin L, Guarnieri G, Grassi G. Inhibitory effects of fenofibrate on apoptosis and cell proliferation in human endothelial cells in high glucose. J Mol Med (Berl) 2008;86:185–195. doi: 10.1007/s00109-007-0257-3. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura J, Dewa Y, Muguruma M, Kuroiwa Y, Yasuno H, Shima T, Jin M, Takahashi M, Umemura T, Mitsumori K. Effect of fenofibrate on oxidative DNA damage and on gene expression related to cell proliferation and apoptosis in rats. Toxicol Sci. 2007;97:44–54. doi: 10.1093/toxsci/kfm011. [DOI] [PubMed] [Google Scholar]
- 29.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 30.Kamijo Y, Hora K, Tanaka N, Usuda N, Kiyosawa K, Nakajima T, Gonzalez FJ, Aoyama T. Identification of functions of peroxisome proliferator-activated receptor α in proximal tubules. J Am Soc Nephrol. 2002;13:1691–1702. doi: 10.1097/01.asn.0000018403.61042.56. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K, Kamijo Y, Hora K, Hashimoto K, Higuchi M, Nakajima T, Ehara T, Shigematsu H, Gonzalez FJ, Aoyama T. Pretreatment by low-dose fibrates protects against acute free fatty acid-induced renal tubule toxicity by counteracting PPARalpha deterioration. Toxicol Appl Pharmacol. 2011;252:237–249. doi: 10.1016/j.taap.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smeets PJ, Teunissen BE, Planavila A, de Vogel-van den Bosch H, Willemsen PH, van der Vusse GJ, van Bilsen M. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem. 2008;283:29109–29118. doi: 10.1074/jbc.M802143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Emmett N, Mann D, Zhao X. Fenofibrate attenuates tubulointerstitial fibrosis and inflammation through suppression of nuclear factor-kappaB and transforming growth factor-beta1/Smad3 in diabetic nephropathy. Exp Biol Med (Maywood) 2010;235:383–391. doi: 10.1258/ebm.2009.009218. [DOI] [PMC free article] [PubMed] [Google Scholar]