

Abstract
Autism is a neurodevelopmental disorder characterized by abnormal reciprocal social interactions, communication deficits, and repetitive behaviors with restricted interests. Most of the available research on autism is focused on children and young adults and little is known about the pathological alternation of autism in older adults. In order to investigate the neurobiological alternation of autism in old age stage, we compared the morphology and synaptic function of excitatory synapses between the BTBR mice with low level sociability and B6 mice with high level sociability. The results revealed that the number of excitatory synapse colocalized with pre- and post-synaptic marker was not different between aged BTBR and B6 mice. The aged BTBR mice had a normal structure of dendritic spine and the expression of Shank3 protein in the brain as well as that in B6 mice. The baseline and KCl-evoked glutamate release from the cortical synaptoneurosome in aged BTBR mice was lower than that in aged B6 mice. Overall, the data indicate that there is a link between disturbances of the glutamate transmission and autism. These findings provide new evidences for the hypothesis of excitation/inhibition imbalance in autism. Further work is required to determine the cause of this putative abnormality.
Keywords: Autism, BTBR mice, aging, excitatory synapse, glutamate release
Introduction
Autism is a severe neurodevelopmental disorder characterized by impairments in social interaction, deficits in verbal and non-verbal communication and repetitive behavior and restricted interests, yet little is known about older adults with autism [1]. The most relevant research indicates that autism is just as prevalent in older people as they are in younger age groups, around 1 in 150 older people is likely to be affected by autism [2]. Studies that investigate the pathology of autism in an aged physiological context are lacking. Animal models are a useful tool in the search for pharmacological treatment for the core symptoms of autism. Inbred strains of mice differ in their levels of sociability, and several strains have reduced social interactions [3,4]. The BTBR T + Itpr3tf (BTBR) mice have emerged as strong candidates to serve as models of a range of autism-relevant behaviors, showing deficiencies in social behaviors and reduced or unusual ultrasonic vocalizations as well as increased repetitive self-grooming [5-7]. Recently, it has been reported that a reduction in social behavior persists into old age in male BTBR mice [8]. However, the neurobiological factors that account for the BTBR sociability levels are largely unknown.
The brains of individuals with autism involve deficits in information processing, characterized by hyperconnectivity in local circuits and hypoconnectivity between brain regions [9]. Inhibitory and excitatory synapses play fundamental roles in information processing in the brain. Inappropriate loss of synaptic stability could lead to the disruption of neuronal circuits and to brain dysfunction [10,11]. BTBR mice in young adults had reduced spontaneous γ-aminobutyric acid (GABA) neurotransmission. Treatment with low doses of benzodiazepines, which increase inhibitory neurotransmission through positive allosteric modulation of postsynaptic GABAA receptors, improved deficits in social interaction, repetitive behavior, and spatial learning [7]. Similarly, another study showed that BTBR mice exhibited weakened GABA circuits and compromised postnatal pruning of cross-modal input. Transient pharmacological enhancement by diazepam in BTBR mice during an early sensitive period rescued inhibition and integration in the adult insular cortex [12]. Nevertheless, the features of brain structure and function in older adults are poorly understood.
The capacity of excitatory synapses to rapidly modify the membrane expression of glutamate receptors plays a critical role in information processing [13]. Most glutamatergic excitatory synapses occur on small protrusions along dendrites called dendritic spines that contain the postsynaptic machinery. Dendritic spines are the major site for excitatory transmission in the brain. Dendritic spine pathology accompany synapse formation, maintenance and elimination, allowing the establishment and remodeling of connectivity within neuronal circuits [14]. Shank family proteins are synaptic scaffold proteins at the postsynaptic density (PSD) of excitatory synapses. Shank3 was the first gene in the Shank family reported to be associated with autism. In vitro and in vivo studies highlight the important role of Shank3 for synaptic function in spine morphogenesis and synaptic plasticity [15].
Glutamate is the principal excitatory neurotransmitter in the central nervous system and plays a critical role in numerous functions, such as cognition, movement, learning and memory [16]. Excessive glutamate release and activation of the glutamate receptors has been implicated as a pathophysiological factor in multiple neurological disorders [17]. However, the role of glutamate release in autism in older adults is unclear. To investigate the neurobiological alternation of autism in old age stage, in the present study, we compared the morphology and synaptic function of excitatory synapses between the BTBR mice with low level sociability and C57BL/6 (B6) mice with high level sociability.
Materials and methods
Animals
B6 and BTBR mice breeding pairs were purchased from Model Animal Research Center of Nanjing University (Nanjing, China) and bred at Shanxi Medical University. Subject mice were weaned at 21±1 days of age, then group housed by sex and strain in standard mouse cages containing 2-4 mice. Standard rodent chow and tap water were available ad libitum. The colony room was maintained on a 12:12 light/dark cycle, with lights on at 7:00 AM. Male BTBR mice (15 months of age) and male B6 mice (15 months of age) were used. We certify that all applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research.
Synaptic assay
For excitatory synapse quantification, animals were sacrificed by decapitation and rapidly the brains were fixed in 4% PFA by direct immersion overnight at 4°C. Then the brains were coronally sectioned (200 μm thick) using a vibratome at room temperature and stored in antifreeze solution (30% ethylene glycol, 20% glycerol, and 50% 0.05 M phosphate buffer) at -20°C until use.
Synapse quantification was performed as previously described [18]. Briefly, sections were washed three times for 10 min in PBS and then placed in 0.01% Triton X-100 in PBS solution for 15 min at room temperature and incubated in blocking solution containing 10% goat serum for 1 h at room temperature. Rabbit anti-Shank3 (1:250, Synaptic Systems) to label glutamatergic postsynaptic compartments and guinea pig anti-VGLUT 1 (1:500, Synaptic Systems) to label glutamatergic presynaptic terminals were added to the PBS, and sections were incubated overnight at 4°C. For some sections, the primary antibody was omitted as a control. Tissue was washed three times for 15 min in PBS, and was incubated in secondary antibody solution consisting of goat anti guinea-pig Alexa Fluor 488 (VGLUT1, 1:1000, Invitrogen) and goat anti rabbit Alexa Fluor 647 (Shank3, 1:1000, Invitrogen) for 1 h at room temperature in the dark. The sections were washed five times in PBS and incubated in DAPI solution. Slices were washed in PBS and mounted with ProLong Antifade (Invitrogen).
Images were collected using a 60× oil-immersion objective lens on a confocal laser scanning microscopy (FV1000, Olympus). As previously described [18], at least 3 animals were included for each group. Synapses were quantified using single confocal planes collected with a 4 × digital zoom factor in somatosensory cortex. Colocalized puncta were identified using the Puncta Analyzer plug-in in NIH Image/J as described [19].
Dendritic spine assay
According to the method described in our previous work [20], the brains were fixed and coronally sectioned (200 μm thick) using a vibratome at room temperature. Slices were incubated with Vybrant-DiI cell-labeling solution (1:150, Invitrogen) for 36 h at 4°C to allow DiI crystals to diffuse fully along the neuronal membranes. Then the slices were bathed in the PBS for 48 h to allow more time for diffusion and mounted on glassslides with ProLong Gold antifade reagent (Invitrogen). All images were taken within 7 days after coverslipping using a 60× oil-immersion objective lens with a 4× digital zoom factor in somatosensory cortex on a confocal laser scanning microscopy (FV1000, Olympus).
Western blot
The mouse cortices were homogenized in RIPA buffer. A Bradford assay was performed to calculate protein yield and 50 μg of the homogenate was resolved via electrophoresis on 10% SDS-PAGE gels. The gels were transferred to PVDF membranes, and incubated in a solution of 5% non-fat milk for 2 h at room temperature. Blots were incubated with anti-Shank3 (1:1000, Synaptic Systems) overnight at 4°C, washed four times for 15 min in PBS with 0.1% Tween 20, followed by 1 h incubation with a horseradish peroxidase conjugated goat anti-rabbit (1:20 000, EarthOx). Blots then were washed four times for 15 min and visualized using chemiluminescent substrate (Millipore). As previously described [8], 3 animals were included for each group. Quantification of band intensity was performed using ImageJ (NIH) and presented relative to the loading control (Actin).
Isolation of synaptoneurosomes and characterization by electron microscopy
Synaptoneurosomes were prepared from mouse brain cerebral cortex as described previously [19,21]. Briefly, whole cerebral cortex was homogenized in synaptoneurosome buffer (118.5 mM NaCl, 4.7 mM KCl, 1 mM CaCl2, 1.18 mM MgSO4, 20 mM HEPES, 9 mM Tris, 10 mM Glucose) at 4°C using a Teflon-glass mechanical tissue grinder (0.25 mm clearance). From this step forward the homogenate was kept ice-cold at all times to minimize proteolysis throughout the isolation procedure. The sample was loaded through three layers of a pre-wetted 147 μm pore nylon filter. The resulting filtrate was placed in a 50 ml polycarbonate tube and centrifuged at 1000× g for 10 min. The pellet obtained corresponded to the synaptoneurosome fraction. The pellet was spun briefly before it was fixed in 2.5% glutaraldehyde for 2.5 h on ice. Samples were washed twice for 10 min with 0.1 M sodium cacodylate, fixed for 2 h in 1% OsO4. Samples were gradually dehydrated with a 5 to 10 min wash of 30-100% acetone. Epoxy resin 618 was used to embed the samples and cured at 55°C for 2-3 days. Five hundred Å thin sections were cut using LKB ultramicrotome and then stained in uranyl acetate and lead citrate. Sections were viewed on JEOL-1101 Transmission Electron Microscope and images were acquired with iTEM image analysis platform.
Glutamate release assay
The glutamate release assay was performed using enzyme-linked fluorescent detection of released glutamate [22,23]. In brief, synaptoneurosomes were stored on ice and diluted to 2 mg/ml protein using Krebs-like solution as described. For each well of a 96 well plate 25 μl synaptoneurosome solution was added to 175 μl calcium-free Krebs-like solution (118 mM NaCl, 5 mM KCl, 25 mM NaHCO3, 1 mM MgCl2, 10 mM Glucose, pH 7.4). After 60 s, 1 mM NADP+ (Sigma) was added ±1.2 mM calcium chloride as required. After ~6 min, five units of glutamate dehydrogenase (Sigma) were added and allowed to equilibrate inside the instrument for at least 10 min prior to the addition of any test drug or depolarisation stimulus. Fluorescence at 460 nm emission was continuously measured using a BioTek Synergy 4 Microplate reader set at the following parameters: 80-120 cycles (number of times a complete plate is read), 15 s cycle time (time between each cycle), FlashMode (high sensitivity), excitation filter 360-40, emission filter 460-40, injection cycle 60, shaking after each cycle for 1 s, 37°C. Depolarisation stimulus (30 mM KCl) and glutamate standards were added by pausing the instrument. Glutamate was quantitated by the addition of 5 nmol glutamate (Sigma) at the end of each run as an internal calibration control.
Statistical analysis
All data were analyzed using commercially available statistical software packages (StatView 5.0 and GraphPad Prism 5). The glutamate release data was analyzed by two-way ANOVA with repeated measures with Bonferroni’s post hoc analysis. The unpaired t-test (two-tailed) was conducted to determine the significant differences for the synaptic assay and Western blot data. The data was shown as mean ± SEM. For all findings, the statistically significant P values are shown as *P<0.05, **P<0.01.
Results
The number of excitatory synapse
Autism is likely to arise from functional changes in neural circuitry and to be associated with an imbalance between excitatory and inhibitory synaptic transmission. However, the distribution of synaptic structure of autism in old age stage is unclear. Because of the close relationship between excitatory synapse and dendritic spine, the present study focused on the change of the excitatory synapse. To investigate the number of the excitatory synapse in the brain of the aged BTBR mice, we double labeled sections of 15-months old BTBR mice and B6 mice using antibodies to the pre-synaptic protein VGLUT1 and the post-synaptic protein Shank3 (Figure 1A). By image analysis, we observed a similar number of colocalized pre- and post-synaptic puncta in BTBR brains compared to B6 (P>0.05, Figure 1B).
Figure 1.
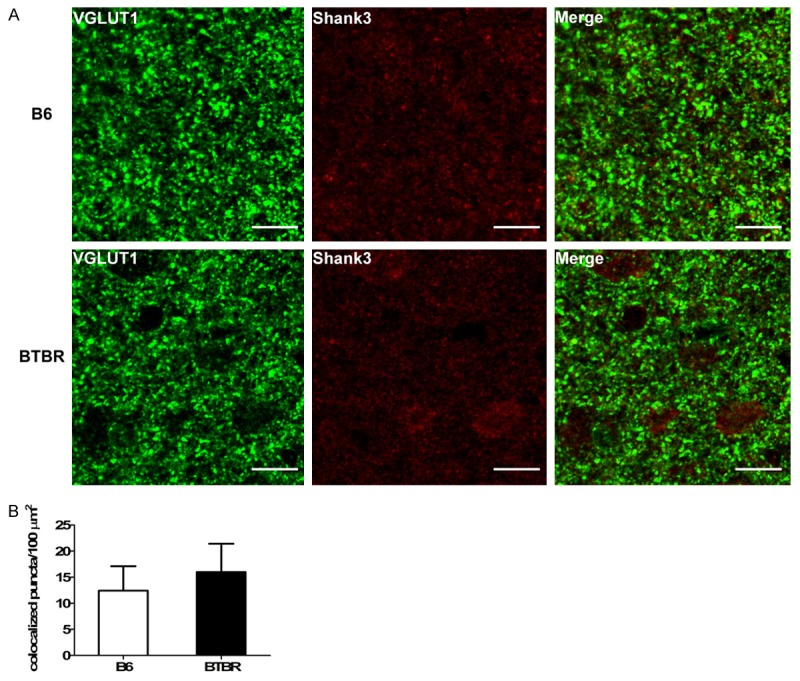
Quantification of colocalization of pre- and post-synaptic puncta. A. Representative immunofluorescence images of B6 and BTBR cortex immunolabeled for pre-synaptic VGLUT1 and post-synaptic Shank3. Scale bar, 10 µm. B. Quantification of colocalization of pre- and post-synaptic markers in brain sections. No difference was observed between B6 and BTBR brains (P>0.05). Data are shown as mean ± SEM.
The shape of dendritic spine
The plasticity of the dendritic spine has been proved to be involved in the pathology of autism. Then, we employed DiI labeling to outline dendritic spines in pyramidal neurons of the somatosensory cortex (Figure 2). The morphology of spine in BTBR cortex was mature and similar to that in the B6 mice. The three members of the Shank family are core components of the postsynaptic density. Shank proteins and their binding partners are involved in the regulation of the size and the shape of dendritic spine [24]. We compared the expression of Shank3 protein in BTBR cortex and B6 cortex using Western blot (Figure 3A) and found no differences between them (P>0.05) (Figure 3B).
Figure 2.
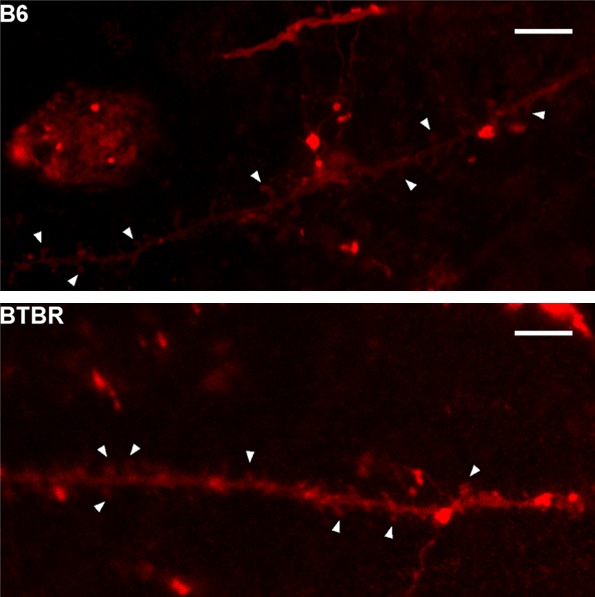
The shape of dendritic spine in B6 and BTBR mice. Confocal micrographs of DiI labeled dendrites in cortex somatosensory layer from B6 mice and BTBR mice. Arrowhead shows the spines. Scale bar, 5 µm.
Figure 3.
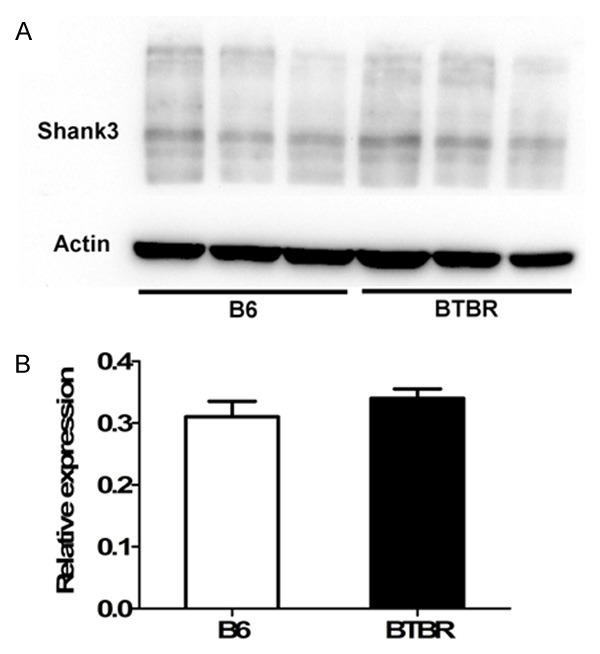
Shank3 protein expression in B6 and BTBR mice. A. Western blot of mouse cortex lysates using Shank3 antibody. B. The blots shown in A were quantitated after normalization by actin respectively. Data are shown as mean ± SEM.
Glutamate release assay of synaptoneurosomes
The excitatory synapses consist of post-synaptic glutamate receptors and pre-synaptic glutamate localized inside vesicles. Glutamate binds to glutamate receptors, giving rise to synaptic transmission. The measurement of glutamate release at synaptic sites provides valuable information to how the synapses function during synaptic transmission. The synaptic junction consists of regions of the presynaptic and postsynaptic plasma membranes joined together by structures within the synaptic cleft. The synaptoneurosome is suggested for entities in which a presynaptic sac (synaptosome) is attached to a resealed postsynaptic sac (neurosome) [25] and can be used for the neurotransmitter assays. In this study, electron micrographs of isolated synaptoneurosome showed a dense cellular mass in which mitochondria and vesicular structures are seen together with densely stained membranes representing PSDs (Figure 4A and 4B). These electron micrographs are in agreement with previous studies [25,26].
Figure 4.
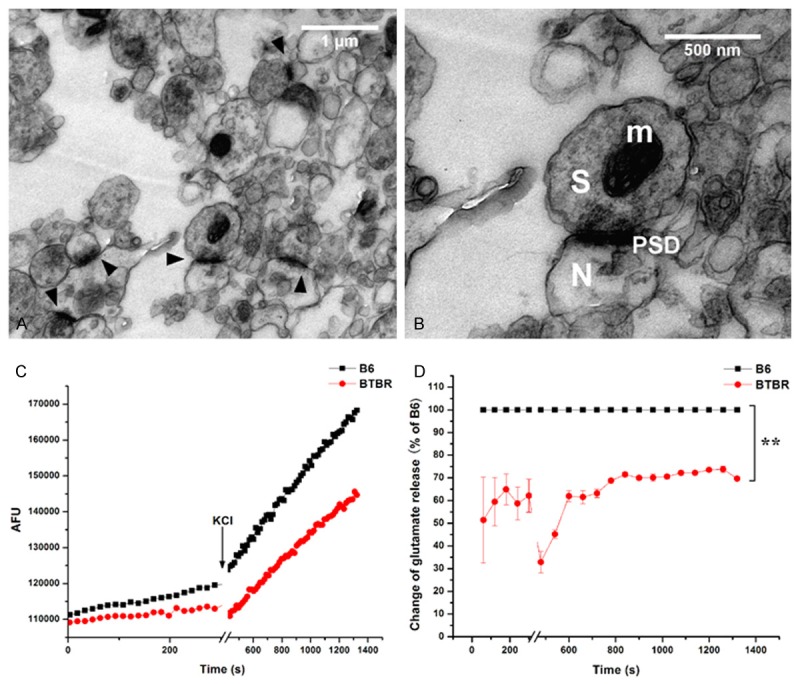
Glutamate release of synaptoneurosomes. A. Electron micrographs of representative sample of synaptoneurosomes isolated from BTBR mouse cortex. Arrowhead indicates synaptic junctions. B. Higher magnification of the synaptic site where the pre-synaptic terminal displays vesicles and a post-synaptic membrane contains PSDs. Synaptosome (S), neurosome (N), and densely stained membranes that characterize PSDs (PSD) are shown. Mitochondria (m) also are present. C. A typical experimental trace for glutamate release each assay is shown. AFU: arbitrary fluorescence units. D. Relative comparison of glutamate release in synaptoneurosomes between B6 and BTBR mice. **P<0.01.
Next, we measured the baseline and evoked endogenous glutamate release using enzyme linked fluorescence detection, with glutamate dehydrogenase and the reduction of NADP to NADPH. Figure 4C showed a typical kinetic trace, consistent with the previous results [22]. On the baseline, the endogenous glutamate release from the synaptoneurosome in BTBR mice was lower than that in B6 mice (P<0.01, Figure 4D). Furthermore, after depolarizated with KCl, the synaptoneurosome in BTBR mice showed an increased glutamate release compared to the baseline, but still significantly lower than the induced-increase in B6 mice (P<0.01, Figure 4D).
Discussion
Most of the available research on autism is focused on children and young adults and little is known about the pathological alternation of autism in older adults. Whether the neurobiological findings in young individuals with autism are present in older persons with an autism is unknown. Studies of aging in animal models of autism may provide insights into the effect of aging on the nervous system in individuals with autism [1]. In this study, we explored the number and neurotransmission of excitatory synapse in brain used an aged BTBR mouse model of autism.
This study showed that the number of excitatory synapse colocalized with pre and post-synaptic markers was not different between aged BTBR and B6 mice. Developmental alterations of excitatory synapses are implicated in autism. Recently, one postmortem study showed that people with autism had an abundance of excitatory synapses in the cerebral cortex. The brain’s normal process of “pruning” bursts of excessive neural synapses that occur during development is impaired in children and adolescents with autism [27]. To date however, none of synaptic studies have been carried out in autism in older adults. In a recent study, Jasien et al. detected the expression of synaptic proteins that regulate aging in the aged BTBR mice [8]. Similarly, expressions of both synaptophysin and PSD95 in cortical tissue were not affected in BTBR mice. Synaptophysin occurs in presynaptic vesicles of neurons and PSD95 localizes at the postsynaptic membrane of excitatory synapses [28]. In accordance with the cortical data they found no significant BTBR-induced changes in the hippocampal expression of synaptophysin or PSD95, although several synaptic proteins were found to be changed using iTRAQ method. These data suggest that, as compared to the B6 mice with high sociability, BTBR with low sociability in old age have the same number of excitatory synapse.
Another piece of evidence is that the aged BTBR mice have the same structure of dendritic spine in the brain as B6 mice. It has been proposed that dendritic spines represent the primary site of structural plasticity in the adult brain, and aging is associated with alterations of dendritic spine morphology and plasticity in prefrontal cortex [29]. The alterations in spine morphology have been linked to the impairments in memory and cognitive functions in aging [30]. We speculate that the low sociability in aged BTBR mice may not correlated with the spine morphology, at least such influence is minimal. Shank3 mutant mice lead to deficits in social interaction and social communication [31,32]. However, this study found no expression of BTBR-related Shank3 protein. Individual genes, such as Shank3, Neuroligin4 and Neurexin1, have been identified to be involved in autism. However, variants at these and other loci are present in no more than 1% to 2% of children with autism [33]. And the existing findings show that autism is not a single clinical entity but a behavioral manifestation of tens or perhaps hundreds of genetic and genomic disorders [34]. Based on the above data and analysis, we suppose that the low sociability in aged autism-like mice may be not due to the change of the number and structure of excitatory synapses.
The most exciting finding in the present study is that the function of glutamate release of excitatory synapse in aged BTBR mice is abnormal. Glutamate is the most important excitatory neurotransmitter in the brain. There is a cumulating body of evidences suggest that the glutamate system exerts an important role in regulating the balance between neuronal excitation and inhibition. Recently, a magnetic resonance spectroscopy study showed a significant decrease in the combined glutamate and glutamine signal (Glx) in anterior cingulate cortex in adults (mean age, 35 years) with high-functioning autism [35]. This signal correlated significantly with impairment in social communication of autism. And it has been reported that individuals with autism (mean age, 29 years) had a significant decrease in concentration of Glx in the basal ganglia [36]. These data support the idea that autism is a hypoglutamatergic disorder. In contrast, Bejjani et al. reported elevated Glx signals in the anterior cingulate cortex in children and adolescents with autism (mean age, 10 years) [37]. And increased frequencies and amplitudes of spontaneous and action-potential-evoked excitatory synaptic potentials have been observed in mouse models of autism bearing human mutations in genes. Accordingly, these evidences point out a hyperglutamatergic hypothesis of autism. As the theory of age-specific anatomic abnormalities in autism states, early brain overgrowth during infancy and the toddler years, followed by an accelerated rate of decline in size and perhaps degeneration from adolescence to late middle age in autism [38,39]. Based on the existing findings, it seems that there is an age-related glutamate changes, excessive glutamate (over-activation) in the autistic brain in young age and deficient glutamate (over-inhibition) in autistic brain in old age. These findings provide new evidences for the hypothesis of excitation/inhibition imbalance in autism [35,40,41].
The limitation of this study is that it is not a longitudinal data to observe the change of excitatory synapse in autism-like mice. However, by compared to an aged mouse that displays relatively normal levels of social behavior, we can get some clues and help in understanding the aging and autism. In conclusion, our data support the hypothesis that there is a link between disturbances of the glutamate transmission and autism. Targeting glutamate metabolism might be a possible approach for the treatment of autism. Further work is required to determine the cause of this putative abnormality. Given the complexity of the autism, it is conceivable that the impairment in neurotransmission of excitatory synapse is unlikely the only pathological mechanism behind autism in older adults.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81201061), Outstanding Youth Talents Program of Shanxi Province, the Ministry of Human Resources and Social Security of China (2014), Shanxi Scholarship Council of China (No. 2013-124) and Natural Science Foundation of Shanxi (No. 2013021036-2).
Disclosure of conflict of interest
None.
References
- 1.Piven J, Rabins P Autism-in-Older Adults Working Group. Autism spectrum disorders in older adults: toward defining a research agenda. J Am Geriatr Soc. 2011;59:2151–2155. doi: 10.1111/j.1532-5415.2011.03632.x. [DOI] [PubMed] [Google Scholar]
- 2.Brugha T, McManus S, Meltzer H, Smith J, Scott F, Purdon S, Harris J, Bankart J. Autism spectrum disorders in adults living in households throughout England-2007, report from the adult psychiatric morbidity survey. United Kingdom: NHS The Information Centre for Health and Social Care; 2009. [Google Scholar]
- 3.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 4.Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, Magnuson TR, Crawley JN. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res. 2007;176:4–20. doi: 10.1016/j.bbr.2006.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyza KZ, Defensor EB, Jensen AL, Corley MJ, Pearson BL, Pobbe RL, Bolivar VJ, Blanchard DC, Blanchard RJ. The BTBR T+ tf/J mouse model for autism spectrum disorders-in search of biomarkers. Behav Brain Res. 2013;251:25–34. doi: 10.1016/j.bbr.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman JL, Tolu SS, Barkan CL, Crawley JN. Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology. 2010;35:976–989. doi: 10.1038/npp.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han S, Tai C, Jones CJ, Scheuer T, Catterall WA. Enhancement of inhibitory neurotransmission by GABAA receptors having alpha2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron. 2014;81:1282–1289. doi: 10.1016/j.neuron.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasien JM, Daimon CM, Wang R, Shapiro BK, Martin B, Maudsley S. The effects of aging on the BTBR mouse model of autism spectrum disorder. Front Aging Neurosci. 2014;6:225. doi: 10.3389/fnagi.2014.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 11.Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- 12.Gogolla N, Takesian AE, Feng G, Fagiolini M, Hensch TK. Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron. 2014;83:894–905. doi: 10.1016/j.neuron.2014.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernardinelli Y, Nikonenko I, Muller D. Structural plasticity: mechanisms and contribution to developmental psychiatric disorders. Front Neuroanat. 2014;8:123. doi: 10.3389/fnana.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Yu S, Fu Y, Li X. Synaptic proteins and receptors defects in autism spectrum disorders. Front Cell Neurosci. 2014;8:276. doi: 10.3389/fncel.2014.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenamyre JT, Porter RH. Anatomy and physiology of glutamate in the CNS. Neurology. 1994;44:S7–13. [PubMed] [Google Scholar]
- 17.Meldrum BS. The role of glutamate in epilepsy and other CNS disorders. Neurology. 1994;44:S14–23. [PubMed] [Google Scholar]
- 18.Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. 2010 doi: 10.3791/2270. pii: 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 20.Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, Li X. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim Biophys Acta. 2012;1822:831–842. doi: 10.1016/j.bbadis.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz-Bloom RD, Engblom AC, Akerman KE, Inglefield JR. Measurement of chloride movement in neuronal preparations. Curr Protoc Neurosci. 2001;7:7–10. doi: 10.1002/0471142301.ns0710s04. [DOI] [PubMed] [Google Scholar]
- 22.Nicholls DG, Sihra TS, Sanchez-Prieto J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J Neurochem. 1987;49:50–57. doi: 10.1111/j.1471-4159.1987.tb03393.x. [DOI] [PubMed] [Google Scholar]
- 23.Sim AT, Herd L, Proctor DT, Baldwin ML, Meunier FA, Rostas JA. High throughput analysis of endogenous glutamate release using a fluorescence plate reader. J Neurosci Methods. 2006;153:43–47. doi: 10.1016/j.jneumeth.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17:71–84. doi: 10.1038/mp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3’:5’-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villasana LE, Klann E, Tejada-Simon MV. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods. 2006;158:30–36. doi: 10.1016/j.jneumeth.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–1143. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glantz LA, Gilmore JH, Hamer RM, Lieberman JA, Jarskog LF. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience. 2007;149:582–591. doi: 10.1016/j.neuroscience.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, McEwen BS, Morrison JH. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. 2011;31:7831–7839. doi: 10.1523/JNEUROSCI.0839-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloss EB, Morrison JH, Hof PR, Dickstein DL. Influence of aging and neurodegeneration on dendritic spine morphology. Translat Neurosci. 2011;2:49–60. [Google Scholar]
- 31.Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrahams BS, Geschwind DH. Connecting genes to brain in the autism spectrum disorders. Arch Neurol. 2010;67:395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 35.Tebartz van Elst L, Maier S, Fangmeier T, Endres D, Mueller GT, Nickel K, Ebert D, Lange T, Hennig J, Biscaldi M, Riedel A, Perlov E. Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol Psychiatry. 2014;19:1314–1325. doi: 10.1038/mp.2014.62. [DOI] [PubMed] [Google Scholar]
- 36.Horder J, Lavender T, Mendez MA, O’Gorman R, Daly E, Craig MC, Lythgoe DJ, Barker GJ, Murphy DG. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [(1)H] MRS study. Transl Psychiatry. 2013;3:e279. doi: 10.1038/tp.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, Kitchen C, Salamon N, McCracken JT, Toga AW, Alger JR, Levitt JG. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS One. 2012;7:e38786. doi: 10.1371/journal.pone.0038786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courchesne E, Campbell K, Solso S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res. 2011;1380:138–145. doi: 10.1016/j.brainres.2010.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- 40.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]