

Abstract
Histone deacetylase HDAC2 regulates genes transcription via removing the acetyl group from histones. Glucocorticoids, the most potent anti-inflammatory treatment available for inflammatory diseases, inhibit the expression of inflammatory genes by recruiting HDAC2 to activated genes. In the lungs of patients who smoke and have chronic obstructive pulmonary disease (COPD) or asthma, glucocorticoids are not effective enough to suppress airway inflammation, which is so called “glucocorticoid resistance”, due to decreased HDAC2 level caused by cigarette smoke. We report that the ubiquitin-specific protease USP17 interacts with HDAC2. USP17 deubiquitinates and stabilizes the protein level of HDAC2. In cigarette smoke extract-exposed airway epithelial cells and macrophages, HDAC2 is excessively ubiquitinated and degraded in the proteasome attributed to low expression of USP17. Furthermore, over-expression of USP17 blocks the destruction of HDAC2 induced by cigarette smoke extract. These results provide a new insight into the mechanisms of glucocorticoid resistance in airway inflammatory disease. Small molecules which can specifically induce the expression of USP17 might be useful in reversing glucocorticoid resistance.
Keywords: HDAC2, USP17, deubiquitination, cigarette smoke extract, glucocorticoid resistance
Introduction
Gene transcription is regulated by the post-translational modification of histones, such as methylation, phosphorylation and acetylation. Through acetylating core histones and subsequent chromatin remodeling, histone acetylases (HATs) allow access of transcription factors and RNA polymerase II to DNA, leading to gene transcription. Meanwhile, histone deacetylases (HDACs) act as a negative regulator of this process, shutting off gene transcription by deacetylation [1]. HDAC2, a member of class I HDAC family, plays a critical role in the regulation of inflammatory genes and in mediating the anti-inflammatory effects of glucocorticoids in asthma and COPD [2].
Glucocorticoids exert their anti-inflammatory effects mainly through directly inhibiting the transcriptional factors-associated HATs and recruiting HDAC2 to activated inflammatory genes [3]. The deacetylation of glucocorticoid receptors (GRs) by HDAC2 has been proved to be a prerequisite for glucocorticoids to switch off activated inflammatory genes, such as nuclear factor-kappaB (NF-ΚB) mediated genes [4]. Various molecular mechanisms of glucocorticoid resistance have been identified, including GR modification, increased GR-β expression, increased proinflammatory transcription factors and reduced HDAC2 expression level and activity [5]. In COPD, severe asthma or smoking asthmatics, who responded poorly to the glucocorticoid treatment, HDAC2 protein expression and activity are remarkably reduced in their alveolar macrophages, bronchial biopsies and peripheral lung tissues [6-8]. The inactivation of HDAC2 is due to the increased 4-hydroxy-2-nonenal (4-HNE) and nitrotyrosine modification of HDAC2 protein caused by cigarette smoke-induced oxidative stress, which could be reversed by antioxidants, N-acetyl-L-cysteine (NAC) or glutathione monoethyl ester (GSHMEE) [9,10]. While the reduced protein level of HDAC2 was mainly attributed to the enhanced proteasomal degradation of HDAC2 under oxidative stress. Cigarette smoke extract (CSE) exposure in MonoMac6 and in human bronchial epithelial and primary small airway epithelial cells led to phosphorylation of HDAC2 on serine/threonine residues and subsequent proteasomal degradation [11]. Hydrogen peroxide (H2O2), another reagent triggers oxidative stress, induced nitration of HDAC2 and following ubiquitination and proteasomal degradation in A549 cells, which would be inhibited by a proteasome inhibitor, N-Acetyl-Leu-Leu-Nle-CHO (ALLN) [12].
Several enzymes mediating the ubiquitination of HDAC2 have been identified. The HDAC inhibitor valproic acid selectively induces proteasomal degradation of HDAC2 via induction of E2 ubiquitin-conjugating enzyme Ubc8 and the E3 ubiquitin ligase RLIM [13]. A HECT domain ubiquitin ligase, Mule (Mcl-1 ubiquitin ligase E3) specifically targets HDAC2 for ubiquitination and degradation [14]. Curcumin, a dietary polyphenol, restored the HDAC2 activity and protein levels by inhibition of its phosphorylation, carbonylation, uiquitination and following proteasomal degradation, thus, reversed the glucocorticoid resistance in U937-differenciated macrophages exposed to CSE [15]. Our research mainly focused on the deubiquitination of HDAC2 and to identify the related deubiquitinating enzymes to HDAC2.
Materials and methods
Reagents
The antibodies used were as follows: anti-Flag (M2, Sigma), anti-Myc (9E10, Santa Cruz), anti-HA (F-7, Santa Cruz), anti-Ubiquitin (sc-8017, Santa Cruz), anti-GAPDH (1C4, Sungene Biotech), anti-β-actin (KM9001, Sungene Biotech), anti-PARP (436400, Invitrogen), anti-HDAC1 (sc-7872, Santa Cruz), anti-HDAC2 (sc-7899, Santa Cruz), anti-HDAC3 (sc-11417, Santa Cruz), anti-mouse IgG HRP (Promega) and anti-rabbit IgG HRP (Jackson). CHX and MG132 were purchased from Merck. Protein AG-beads were obtained from Santa Cruz.
Plasmids
The Plasmids pIP-Myc-USP2, pIP-Myc-USP3, pIP-Myc-USP4, pIP-Myc-USP5, pIP-Myc-USP7, pIP-Myc-USP10, pIP-Myc-USP12, pIP-Myc-USP14, pIP-Myc-USP17, pIP-Myc-USP18, pIP-Myc-USP21, pIP-Flag-USP22, pIP-Flag-USP44, pIP-Myc-CYLD, pIP-HA-A20, pIP-Myc-YOD1, pIP-Flag-HDAC2, pIP-His-Ubiquitin, pIP-His-K48only Ubiquitin, pIP-His-K63only Ubiquitin, pIP-HA-GFP, pIP-Myc-USP17C89S were constructed and kindly provided by our colleagues, Zhang Jin, Yang Jin and Han Lei.
Cell culture and treatments
Human embryonic kidney cell line (HEK 293T) and human lung adenocarcinoma epithelial cell line (A549) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 u/ml penicillin and 100 mg/ml streptomycin (Gibco) at 37°C and 5% CO2. Cells were transfected with indicated plasmids using polyethylenimine (PEI) (Polysciences) or Lipofectami ne® 2000 (Lipo) (Life Technologies) according to the manufacturer’s instructions. Human acute monocytic leukemia cell line (THP-1) was maintained in complete growth medium (RPMI-1640) supplemented with 10% FBS, 1× non-essential amino acids (NEAA), 1× GlutaMAXTM-I, 100 mM sodium pyruvate, 100 u/ml penicillin and 100 mg/ml streptomycin (Gibco) at 37°C and 5% CO2. THP-1 cells were differentiated into the adherent “macrophage-like” cells after treatment of 100 ng/ml PMA (Sigma) for 12 h.
Preparation of cigarette smoke extract (CSE)
Cigarettes were purchased from Double Happiness (Shanghai). Cigarette smoke extract (CSE) was prepared by bubbling smoke from one cigarette into 10 ml of culture medium containing 0.5% FBS using a CSE-generating apparatus as described previously [11,15,16]. Extract, defined as 10% CSE as stock solution, was freshly prepared and sterile filtered through a 0.22-µm filter for all experiments.
Western blotting and immunoprecipitation
Cells were lysed in RIPA buffer consisting of 50 mM Tris-HCl pH 7.5, 135 mM NaCl, 1 mM EDTA, 0.5% NP-40, 10% Glycerol, 0.25% Na-deoxycholate, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4 and 1% Protease Inhibitor Cocktail (P8340, Sigma), followed by immunoprecipitation with indicated antibodies. For Western blotting, cells were lysed in 2× SDS loading buffer containing 20 mM Tris-HCl pH 8.0, 100 mM DTT, 2% SDS, 20% Glycerol, 0.016% BpB, separated by SDS/PAGE, and analyzed by indicated antibodies.
Ni-NTA pull down assay
The cells were lysed with urea lysis buffer containing 10 mM Tris-HCl pH 8.0, 8 M Urea, 100 mM Na2HPO4, 0.2% Triton X-100, 10 mM Imidazole and fully disrupted by ultrasonication. Then the cell lysate was incubated with Ni-NTA agarose beads for 3 h to get His-tag combined with Ni-NTA. After that, Ni-NTA agarose beads was wash with wash buffer and 2× SDS loading buffer was added to get samples for Western Blotting. The ubiquitination modification was analyzed using indicated antibodies.
Quantitative real-time PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen) and complementary DNA (cDNA) was reverse-transcribed using PrimeScript® RT reagent Kit (Takara) following the manufacturer’s instructions. PCR reactions for detecting human genes were carried out using SYBR green mix (TAKARA) on ABI Prism 7900 Sequence Detection System. Relative mRNA expression of indicated genes was presented as the formula 2-ΔCT normalized to β-actin expression. The sequences of primers used were as follows: USP17-forward: 5’-gagcacttggtggaaagagc-3’ and reverse: 5’-tgatggttcttcatcccaca-3’; HDAC2-forward: 5’-tccaaggacaacagtggtga-3’ and reverse: 5’-tgaagccagaagtcttcaaaaag-3’, β-actin-forward: 5’-ctcttccagccttccttcct-3’ and reverse: 5’-ggcagtgatctccttct-3’.
Statistical analysis
All data are presented as means ± SEM of at least three independent experiments. Comparisons between two groups were analyzed using the Student t test (unpaired, two tailed). P < 0.05 or P < 0.01 is considered significant.
Results
USP17 deubiquitinates and interacts with HDAC2
HDAC2, which can suppress the inflammatory genes via deacetylation of histones and GRs, is ubiquitinated and degraded in response to oxidative or nitrative stress [17]. To date, the E2 Ubc8, E3 RLIM and Mule, which tag HDAC2 to destruction, have been identified [13,14]. Whether HDAC2 is under regulation of the deubiquitinating enzymes (DUBs) is not known yet. We utilized the existing DUBs plasmids and the Ni-NTA pulldown assay to screen the USPs specific to HDAC2. HDAC2 was ubiquitinated in HEK 293T cells, while co-transfection of USP2b (lane 4) or USP17 (lane 12) can significantly reduce the ubiquitination of HDAC2 compared with the positive control (lane 3) (Figure 1A). To verify this result, we performed the co-immunoprecipitation in HEK 293T cells with over-expression of Flag-HDAC2 and Myc-USP17 plasmids. The cell lysate was immunoprecipited using anti-Flag or anti-Myc antibodies. We found that HDAC2 and USP17 interact reciprocally (Figure 1B, 1C). These results suggested that HDAC2 may be the substrate to USP17.
Figure 1.
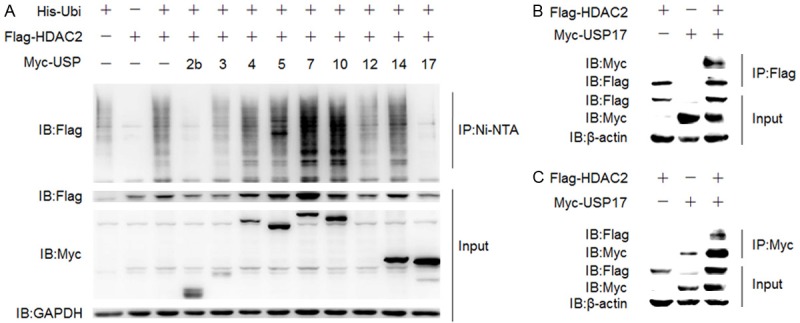
USP17 deubiquitinates and interacts with HDAC2. A. HEK 293T cells were transfected with His-Ubiquitin, Flag-HDAC2, Myc-USP plasmids using PEI and then cultured for 48 h. Before harvested for Ni-NTA pulldown assay, cells were treated with MG132 (10 nM) for 4 h. B and C. HEK 293T cells were transfected with Flag-HDAC2, Myc-USP17 plasmids using PEI and then cultured for 48 h. The cell lysate was immunoprecipitated with anti-Flag (B) or anti-Myc (C) antibody.
USP17 stabilizes HDAC2 by deubiquitination
The ubiquitin molecule contains seven lysine residues at sites 6, 11, 27, 29, 33, 48 and 63. Different types of ubiquitin chains are assembled through isopeptide bonds involving certain lysine of ubiquitin. In general, the canonical lysine 48-linked ubiquitin chains mark proteins to degradation, and other non-canonical ubiquitin chains, like lysine 63-linked ubiquitin chains change the localization or catalytic function of proteins [18]. All lysines other than the lysine at site 48 or 63 of wild type ubiquitin are mutated into alanines to generate the K48 only and K63only mutants. After that, HEK 293T cells were transfected with Flag-HDAC2, His-K48only or K63 only-ubiquitin and Myc-USP17 as shown. Both the K48 and K63-linked ubiquitin chains covalently combined to HDAC2 were removed by USP17 to a certain extent (Figure 2A). As previously discussed, the K48-linked ubiquitin chains lead target proteins to degradation; we speculated that HDAC2 might be stabilized by USP17.
Figure 2.
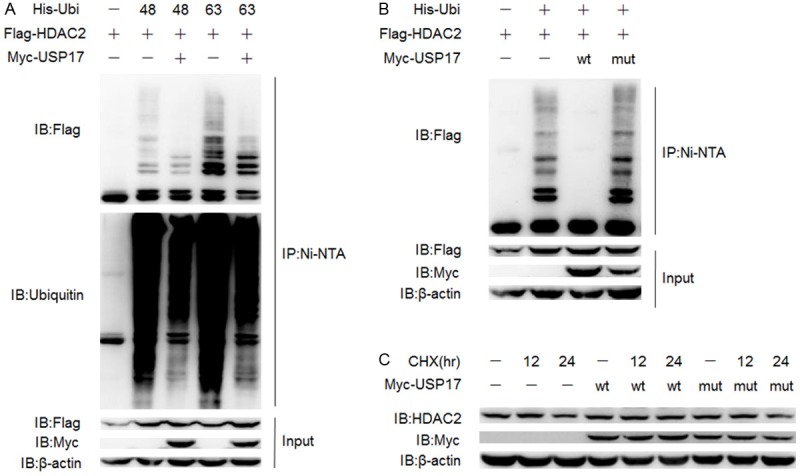
USP17 stabilizes HDAC2 by deubiquitination. A. HEK 293T cells were transfected with Flag-HDAC2, Myc-USP17, His-K48 only or K63 only-Ubiquitin plasmids, lysed for Ni-NTA pull down assay. B. HEK 293T cells were transfected with His-Ubiquitin, Flag-HDAC2, Myc-USP17 or its inactive mutant plasmids using PEI and then cultured for 48 h, lysed for Ni-NTA pull down assay. C. A549 cells were transfected with Myc-USP17 or its inactive mutant plasmids using Lipo and treated with CHX (20 μg/ml). Cells were harvested at indicated time-points and lysed for Western Blotting.
USP17 is proved to inhibit proteasome-mediated degradation of the transcriptional factor retinoic acid-related orphan nuclear receptor gamma t (RORγt) and upregulate Th17-related genes in Th17 cells [19]. To further investigate the effects of USP17 on the expression of HDAC2, the catalytic defective mutant of USP17, Myc-USP17C89S plasmids was adopted in following experiments. Ni-NTA pulldown assay demonstrated that USP17C89S mutant (lane 4) totally lost the deubiquitinating capability towards HDAC2 compared to wild type USP17 (lane 3) (Figure 2B). Then we tested the half-life of endogenous HDAC2 with over-expression of Myc-USP17 or Myc-USP17C89S plasmids in A549 cells in the presence of CHX, a protein synthesis inhibitor. Western Blotting showed that USP17 could prolong the half-life of HDAC2 while the inactive mutant USP17C89S not, which means the enzymatic activity of USP17 was essential for its ability to stabilize HDAC2 (Figure 2C). In contrast to the short half-life of transcriptional factors [19], HDAC2 seemed rather stable in physiological situation.
Over-expression of USP17 inhibited the proteasomal degradation of HDAC2 after CSE exposure
CSE or H2O2 treatment mimicking cigarette smoke exposure in human lungs were reported to induce the ubiquitination and destruction of HDAC2 in the airway epithelial cell lines and macrophage-like cell lines [11,12]. Here we constructed the CSE model in A549 cells to further explore the effects of USP17 mediated-deubiquitination of HDAC2. The A549 cells were treated with various concentrations of CSE and harvested at certain time-points. The protein levels of HDACs were analyzed by Western Blotting. HDAC2 was decreased in both dose-dependent and time-dependent manner. There was no clear change at the protein expression of HDAC1 and HDAC3, another two members belonging to class I HDAC family. The reduction in HDAC2 expression was spontaneously reversed if the A549 cells were exposed to CSE longer than 4 h (Figure 3A). This phenomenon might be caused by the losing efficacy of CSE. Low concentrations of CSE treatment for short time period was not toxic to the A549 cells, as shown by the LDH release, trypan blue exclusion and double staining of acridine orange and ethidium bromide [20]. Here we measured the expression of poly ADP-ribose polymerase (PARP), a marker of cell apoptosis, which can be clove by caspase-3. There was no cleavage of PARP after CSE exposure, indicating no activation of cell apoptosis (Figure 3A). The reduction in HDAC2 protein level was restored by MG132, a proteasome inhibitor (Figure 3B). Besides, the decreased HDAC2 was not due to changes on its transcriptional regulation, as the mRNA level of HDAC2 in CSE-treated A549 cells showed no difference to control group (Figure 3C). Furthermore, CSE treatment induced decreased mRNA expression of USP17 (Figure 3D). From results above, we assumed that CSE treatment tilted the ubiquitination/deubiquitination balance of HDAC2 towards ubiquitination via the downregulation of USP17. To verify our hypothesis, we examined the ubiquitination level of HDAC2 in A549 cells and found that the polyubiquitination of HDAC2 was significantly increased in a dose-dependent manner after CSE treatment (Figure 3E). The over-expression of USP17 reversed the low protein level of HDAC2 induced by CSE treatment (Figure 3F). These results (Figures 2C, 3F) proved that USP17 could stabilize the protein level of HDAC2 under both physiological and pathophysiological conditions.
Figure 3.
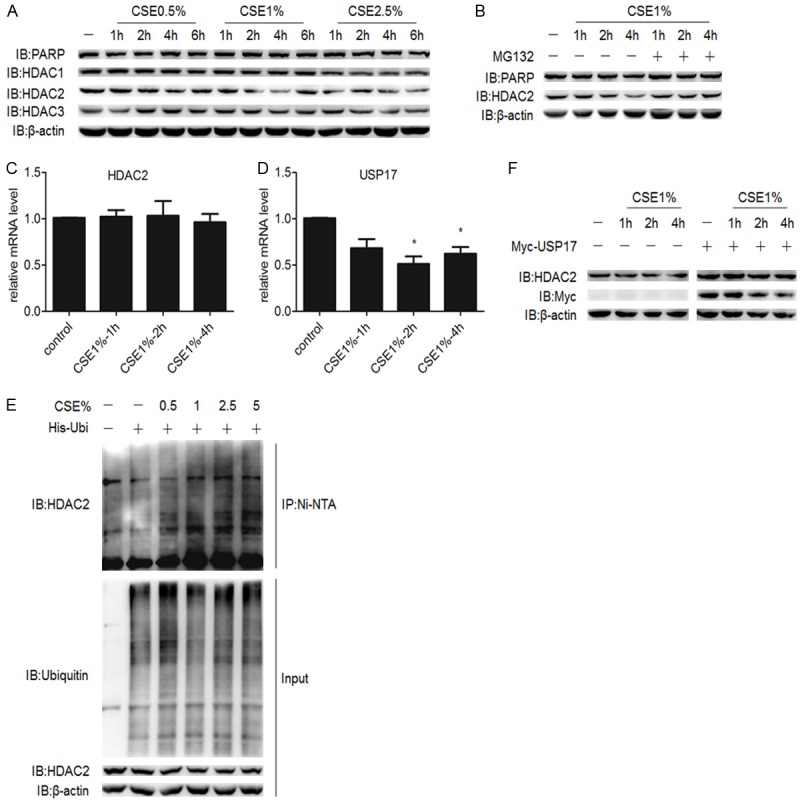
Over-expression of USP17 inhibited the proteasomal degradation of HDAC2 after CSE exposure. A. A549 cells were exposed to indicate concentrations of CSE and harvested at indicated time-points, followed by Western Blotting. B. A549 cells were exposed to CSE and treated with or without MG132 (10 nM) before harvested for Western Blotting. C, D. A549 cells were exposed to CSE and harvested at indicated time-points. The mRNA levels of HDAC2, USP17 were analyzed by q-PCR. The data were plotted as means ± SEM, representing at least three experiments. *P < 0.05 compared with control values. E. A549 cells were transfected with His-Ubiquitin plasmids and cultured for 48 h. Then the cells were exposed to CSE of indicated concentrations and treated with MG132 (10 nM) for 4 h before harvested for Ni-NTA pull down assay. F. A549 cells were transfected with or without Myc-USP17 plasmids and cultured for 48 h. Then the cells were exposed to CSE and harvested at indicated time-points for Western blotting.
Expression of USP17 positively correlates with the expression of HDAC2 in the CSE-exposed macrophages
Macrophages are the most abundant inflammatory cells found in the lungs, participating in airway remodeling and eosinophilic inflammation in asthma [21]. The cytokines and chemokines secreted by macrophages such as IL-1β, IL-6, IL-8 and TNFα help airway epithelial cells to recruit neutrophils and eosinophils [22]. CSE treatment reduced the protein level of HDAC2, along with the activation of NF-ΚB, increased production of IL-8 and TNFα [10]. We first induced the THP-1 cells to differentiate into “macrophge-like” cells using PMA. These macrophages exposed to CSE exhibited a reduction in the protein expression of HDAC2 in addition to the slightly reduced HDAC1 protein level. Inhibition of the proteasome by MG132 treatment blocked the degradation of HDAC2 and restored its protein level (Figure 4A). The mRNA expression of USP17 was similarly decreased after CSE treatment (Figure 4B), suggesting there was a positive correlation between the expression of HDAC2 and USP17 in macrophages.
Figure 4.
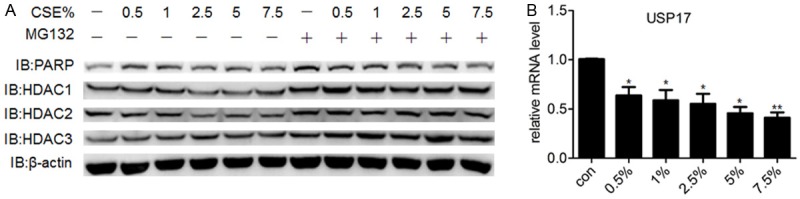
The expression of USP17 positively correlates with the expression of HDAC2 in the CSE-exposed macrophages. A. THP-1 cells were differentiated into “macrophage-like” cells after treatment of 100 ng/ml PMA for 12 h. Then the differentiated macrophages were treated with CSE of different concentrations for 4 h with or without MG132 (10 nM). THP-1 cells were lysed for Western Blotting and the expression level of HDAC1, HDAC2 and HDAC3 were examined using indicated antibodies. B. The differentiated macrophages were exposed to CSE for 4 h and total RNA were extracted for q-PCR. The data were shown as means ± SEM, representing at least three independent experiments. *P < 0.05 and **P < 0.01 compared with control values.
Discussion
The ubiquitin-proteasome system regulates the degradation and function of proteins, thus participating in many cellular processes. The concerted actions of the E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes and the E3 ubiquitin ligases covalently attach the ubiquitin molecules to the target proteins, called “ubiquitination”. The deubiquitinating enzymes negatively regulate this process [23]. Our research discovered the deubiquitinating enzyme to HDAC2, USP17, which can deubiquitinate and stabilize HDAC2. The reduced protein level of HDAC2 is relevant to the downregulation of USP17 in the CSE-exposed airway epithelial cells and macrophages.
HDACs are widely spread in various cell types. Cooperation of HDACs removes acetyl group from core histones re-establishing the packed structure of nucleosome, therefore, leading to trans-repression of the genes [1]. HDACs can deacetylate some non-histone proteins, such as p53 and nuclear receptors [24]. Among them, the interaction of HDAC2 and GR is highly important in the anti-inflammatory effects of glucocorticoids. Upon ligand banding, GRs translocate into nucleus to activate transcription of anti-inflammatory genes, on the other hand, activated GRs recruit HDAC2 binding to inflammatory genes and suppress its expression [3]. The latter one is the major action of glucocorticoids to inhibit inflammation. The GRs are hyper-acetylated to induce transactivation under inflammatory conditions. The application of glucocorticoids enables the deacetylation of GRs by HDAC2 and hypo-acetylation of GRs is demanded for the GRs to stop inflammatory genes expression [4]. Glucocorticoid resistance poses a large obstacle to effective treatment and accounts for considerable health-care expenditure in inflammatory diseases, such as refractory asthma and COPD [5]. In the lungs of these patients, the protein level and activity of HDAC2 is gradually reduced with increasing disease severity and frequent acute exacerbations [7,8]. A common character of these patients is smoking. The relevance of smoking and low HDAC2 expression was supported by later in vivo and in vitro experiments, which proved the proteasomal destruction of HDAC2 induced by cigarette smoke [11,25].
Upregulating the protein expression or activity of HDAC2 to normal levels could strengthen the effectiveness of glucocorticoids. The over-expression of HDAC2 using a plasmid vector would restore glucocorticoid sensitivity in alveolar macrophages from COPD patients [4]. Combination therapy with an inhaled glucocorticoid and low-dose theophylline may attenuate the airway inflammation and improve lung function in patients of COPD. Compared with inhaling glucocorticoid alone, addition of theophylline prominently increased the activity of HDAC2 [26]. In CSE-treated macrophage-like cells, which is resistant to glucocorticoids, curcumin helped glucocorticoids to inhibit the release of cytokines via blocking the degradation of HDAC2 rather than its anti-oxidant property [15]. These results suggested that HDAC2 would be one of the targets in the treatment of glucocorticoid resistance.
Our results found the ubiquitin-specific protease USP17 could increase the protein expression level of HDAC2 by interacting with and deubiquitinating HDAC2. Moreover, in the CSE-induced inflammation, expression of endogenous USP17 declined, resulting in the ubiquitination and construction of HDAC2. And over-expression of USP17 stabilized the protein level of HDAC2 in the CSE-exposed cells, indicating that drugs which can specifically elevate the expression of USP17 may be a promising candidate in the treatment of glucocorticoid resistance attributed to low level of HDAC2. Investigating the changes of USP17 and HDAC2 in the glucocorticoid resistant airway disease and screening small molecules targeting USP17 will be our future work.
Acknowledgements
Our research was supported by the grants from Chinese National Program on Key Basic Research Project 2014CB541800, 2014CB541900, and National Natural Science Foundation of China 81270083, 81470216. We would like to thank all the lab members from Key Laboratory of Molecular Virology and Immunology, Unit of Molecular Immunology, Institute Pasteur of Shanghai for technical assistance. And we would like to express our gratitude to the doctors from Department of Pulmonary Medicine, Rui Jin Hospital for their professional advices.
Disclosure of conflict of interest
None.
References
- 1.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009;3:235–243. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- 3.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi Y, Bossley C, Gupta A, Akashi K, Tsartsali L, Mercado N, Barnes PJ, Bush A, Ito K. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Chest. 2014;145:305–312. doi: 10.1378/chest.13-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 8.Qu Y, Yang Y, Ma D, He L, Xiao W. Expression level of histone deacetylase 2 correlates with occurring of chronic obstructive pulmonary diseases. Mol Biol Rep. 2013;40:3995–4000. doi: 10.1007/s11033-012-2477-z. [DOI] [PubMed] [Google Scholar]
- 9.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004;18:1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 10.Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 11.Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osoata GO, Yamamura S, Ito M, Vuppusetty C, Adcock IM, Barnes PJ, Ito K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem Biophys Res Commun. 2009;384:366–371. doi: 10.1016/j.bbrc.2009.04.128. [DOI] [PubMed] [Google Scholar]
- 13.Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Gottlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. Embo J. 2003;22:3411–3420. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Kan S, Huang B, Hao Z, Mak TW, Zhong Q. Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 2011;25:2610–2618. doi: 10.1101/gad.170605.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, Marwick JA, Chakravarty P, Fletcher D, Whittaker P, Megson IL, Kirkham PA, Rahman I. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol. 2008;39:312–323. doi: 10.1165/rcmb.2008-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu FH, Xiong D, Xu YF, Cao SM, Xue WQ, Qin HD, Liu WS, Cao JY, Zhang Y, Feng QS, Chen LZ, Li MZ, Liu ZW, Liu Q, Hong MH, Shugart YY, Zeng YX, Zeng MS, Jia WH. An epidemiological and molecular study of the relationship between smoking, risk of nasopharyngeal carcinoma, and Epstein-Barr virus activation. J Natl Cancer Inst. 2012;104:1396–1410. doi: 10.1093/jnci/djs320. [DOI] [PubMed] [Google Scholar]
- 17.Barnes PJ. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:693–696. doi: 10.1513/pats.200907-071DP. [DOI] [PubMed] [Google Scholar]
- 18.Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125:539–548. doi: 10.1242/jcs.093567. [DOI] [PubMed] [Google Scholar]
- 19.Han L, Yang J, Wang X, Wu Q, Yin S, Li Z, Zhang J, Xing Y, Chen Z, Tsun A, Li D, Piccioni M, Zhang Y, Guo Q, Jiang L, Bao L, Lv L, Li B. The E3 deubiquitinase USP17 is a positive regulator of retinoic acid-related orphan nuclear receptor gammat (RORgammat) in Th17 cells. J Biol Chem. 2014;289:25546–25555. doi: 10.1074/jbc.M114.565291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. 2012;5:605–609. doi: 10.1038/mi.2012.74. [DOI] [PubMed] [Google Scholar]
- 22.Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy. 2014;69:817–827. doi: 10.1111/all.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clague MJ, Coulson JM, Urbe S. Cellular functions of the DUBs. J Cell Sci. 2012;125:277–286. doi: 10.1242/jcs.090985. [DOI] [PubMed] [Google Scholar]
- 24.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1174–1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
- 26.Ford PA, Durham AL, Russell RE, Gordon F, Adcock IM, Barnes PJ. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 2010;137:1338–1344. doi: 10.1378/chest.09-2363. [DOI] [PubMed] [Google Scholar]