

Abstract
Purpose: MicroRNA-323 (miR-323) has been reported to be upregulated in Ischemia/Reperfusion (I/R) injury-treated neuronal cell. However, the effect and underlying mechanism of miR-323 in I/R-induced neuronal cell death remains poorly understood. The current study was aim to investigate the role and molecular basis of miR-323 in I/R-induced neuronal cell. Methods: An oxygen-glucose deprivation (OGD) model of hippocampal neuron I/R was produced in vitro. Cell apoptosis, cell survival, and the expression of miR-323 were determined after 6 h, 12 h and 24 h after OGD treatment. The up- or down-regulation of miR-323 was performed by miR-323 mimics or anti-miR-323, respectively. Results: OGD induced apoptosis and suppressed survival in rat hippocampal neurons. And the expression levels of miR-323 were increased after OGD treatment. Furthermore, the up-regulation of miR-323 promoted apoptosis and suppressed survival, whereas the inhibition of miR-323 suppressed apoptosis and enhanced survival in OGD-treated neurons. Moreover, miR-323 could directly bind to BRI3 3’-UTR. Notably, the knockdown of BRI3 by BRI3 siRNA apparently abrogated cell survival and induced cell apoptosis in rat neurons. Conclusion: This study indicated that miR-323 might regulate ischemia/reperfusion-induced rat neuronal cell death via targeting BRI3.
Keywords: MicroRNA-323, ischemia/reperfusion injury, neuronal apoptosis, BRI3
Introduction
Stroke is a frequent and often disabling disease, remains a major cause of mortality and morbidity in elderly individuals [1,2]. Ischemic stroke is the most common stroke subtype resulting from an obstruction within a blood vessel supplying blood to the brain [1]. Unfortunately, the therapy of blood flow reperfusion and reoxygenation exacerbates tissue injury [3].
Ischemic stroke and ischemia-reperfusion (I/R) injury can lead to the blood-brain barrier (BBB) destruction and brain vasogenic edema [4,5]. This event accompanied by oxidative stress, inflammation, apoptosis, excitotoxicity, and so on [6]. Oxidative stress caused by post I/R can hinder protein synthesis, causes deleterious DNA mutations, and ultimately leading to apoptosis in neurons [7,8]. Therefore, inhibition neuronal apoptosis after I/R injury is an effective therapy for ischemic stroke. However, the precise mechanism of I/R-induced neuronal death remains poorly understood.
MicroRNAs (miRNAs) are small (~21-nucleotide), noncoding single-stranded RNAs, and more than 2,500 miRNAs molecules have been predicted or verified within human cells [9,10]. Mature miRNAs and Argonaute (Ago) proteins form a ribonucleoprotein complex, the RNA-induced silencing complex (RISC) [11]. Complementary base-pairing of the miRNA guides RISC to target the 3’ untranslated region (3’-UTR) of the messenger RNAs (mRNA), which are degraded, destabilized or translationally inhibited by the Ago protein [12,13]. Proteomic studies have recently uncovered the broad impact of miRNAs on protein output and protein synthesis [14,15]. Up to now, miRNA have become a major focus of research in molecular biology [16,17]. MiRNAs play important roles in a wide range of biological processes such as development, cellular differentiation, proliferation, and apoptosis [12]. Emerging evidence also implicates miRNAs in the pathogenesis of human diseases including cancer and metabolic disorders [12].
Convincing evidences have revealed that a variety of miRNAs were involved and functional in cerebral ischemia reperfusion injury [18]. Altered miRNA expression was detected in the I/R spinal cord. Of these miRNAs, miR-323has been demonstrated to be high expressed in the I/R spinal cord [19]. Thereby, we predicted miR-323 might be implicated in I/R-induced brain injury.
The purpose of the present series of experiments was to characterize the probably role and underlying mechanism of miR-323 (MI0000591) in I/R-induced neuronal apoptosis. The oxygen-glucose deprivation (OGD) model of cell ischemia in vitro was produced to investigate the role of miR-323 in regulating OGD-induced neuron death. Moreover, the underlying mechanism was also investigated using the up- or down-regulation of miR-323 and the target prediction tool.
Material and methods
Cells and cell culture
Rat primary hippocampal neuron cultures were prepared from neonatal SD rats. Briefly, the hippocampi tissues were dissected and dissociated in trypsin-EDTA (0.25%) and primary hippocampal neurons were maintained in neurobasal media (Life Technologies, Carlsbad, CA, USA) in supplement with GlutaMAX and B27 plus glucose (4.5 g/l) for 7 days. Then, cells were cultured in a medium containing 5% horse serum (Sigma-Aldrich, St. Louis, Missouri, USA) and 5% FBS (Sigma) supplemented with 15 mM glucose for 14 days. All cells were cultured in an incubator with a humidified atmosphere of 5% CO2 at 37°C. The culture neurons were used for in vitro studies at day 8.
OGD-treatment of hippocampal neurons
The OGD-treatment of hippocampal neurons was performed according to a previous report, with some modifications [20]. Briefly, the culture medium were replaced with glucose-free DMEM, and cells were cultured in hypoxic conditions (1% O2/94% N2/5% CO2) at 37°C for 3 h. Then the media were discarded and normal DMEM medium containing glucose was added and continued to culture for 6-24 h of reoxygenation under normoxic condition (95% air/5% CO2). Cells cultured in growth culture medium under normoxic condition were used as control.
Cell apoptosis assay
Flow cytometry was used to analyze cell apoptosis, and Annexin V-propidium iodide (AV-PI) staining was performed. Briefly, after treatment, cells were harvested and washed three times with PBS. After centrifuged for 10 min, cells were resuspended in 500 µl of binding buffer including 5 µl FITC-conjugated Annexin V, the mixture was incubated in the dark for 10 min, and then 5 µl of PI was added. Ultimately, all specimens were assessed by flow cytometry with a FACSCalibur using the CellQuest software (BD Biosciences, San Jose, CA, USA), and all the results are shown as a percentage of total cells counted.
Cell survival assay
Cell survival was determined using XTT assay performed according to the manufacturer’s instructions (Roche Applied Sciences, Tokyo, Japan). Cells were seeded into 96-well plates at a density of 2×103 cells per well in 100 µl of culture medium with or without compounds to be tested. Cells were cultured in a CO2 incubator at 37°C for 24-48 h. Ten microliters of XTT mixture was added to each well and mixed gently. Cells were incubated for 2-4 h at 37°C in a CO2 incubator. The absorbance of each sample was measured using a microplate reader (Benchmark Plus, BioRad, Hercules, CA, USA) at a wavelength of 450 nm. Three independent assays were conducted.
Transfection of miRNAs
The miR-323 mimics, miR-323 inhibitor and negative control miRNAs were obtained from GenePharma Co., Ltd (GenePharma, China). For miRNA transfection, cells were seeded into 24-well plates and grown in the antibiotic free medium overnight until 30-50% confluence was reached. About 0.4 nmol microRNAs was mixed with 15 µl lipofectamineTM 2000 transfection reagent, and the cells were maintained in a 37°C, 5% CO2 incubator for 6 h. Then the medium was replaced with fresh medium for 48 h, after which the expression of miR-323 was then confirmed by quantitative PCR.
siRNA transfection
BRI3 siRNA and control siRNA were purchased from Cell Signaling (Beverly, MA). About 5×104 cells were seeded in each cell of the 24-well micro-plates. The cells were then incubated with a mixture of siRNA and Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in 100 μl of serum-free OPTI-MEM according to the manufacturer’s instructions. The transfection efficiency was detected by real-time PCR and Western blot.
Real-time RT-PCR
Total RNA was isolated from cultured cells using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Real-time RT-PCR was performed in a Rotor-Gene RG-3000 Real-Time Thermal Cycler (Corbett Research, Australia). The expression of miR-323 was determined using the miRNA qPCR detection kit (Gene Copoeia, USA) and BRI3 was amplified using a SYBR® Premix Ex Taq™ II kit (Takara, Dalian, China) and gene-specific primers. The relative expression levels for miR-323 and BRI3 mRNA were normalized using the 2-ΔΔCT method [21] relative to small nuclear RNA U6 (U6 snRNA) and β-actin, respectively.
Western blot
The proteins from cells were extracted using RIPA lysis buffer (Beyotime, Nantong, China) and separated by SDS-PAGE and blotted onto a pre-wet nitrocellulose membrane (GE healthcare, Germany). The membranes were blocked in 10% defatted milk in PBS at 4°C for 2 h, and then probed with different primary antibodies. Antibodies used include rabbit polyclonal anti-BRI3 (1:1000, Abcam, Cambridge, UK), mouse monoclonal anti-β-actin and anti-mouse (1:5,000) horseradish peroxidase-conjugated secondary antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The illuminance was scanned using the Typhoon scanner (Amersham Biosciences, Piscataway, NJ, USA). All experiments were performed in triplicate.
Dual-Luciferase reporter assa
The fragment from the 3’-UTR of BRI3 mRNA containing the predicted miR-323-binding sequences was amplified and subcloned into pGL3 luciferase promoter vector (Promega, Madison, WI, USA). The pGL3 vector containing 3’-UTR of CREB mRNA or mutated forms was co-transfected with AAV-pre-miR-323 or controls into HEK293 cells and incubated for 48 h. Then, cells were harvested and lysed, and the luciferase report activity was detected using the dual-luciferase reporter assay kit (Promega) as per standard protocols. The relative luciferase report activities were normalized to that of the negative control cells.
Statistical methods
The statistical analysis software SPSS 19.0 was used for statistical analysis (SPSS, Chicago, IL, USA). Statistical significance was assessed by an unpaired, two-tailed Student t-test for single comparison or ANOVA for multiple comparisons.
Results
OGD/reoxygenation induces neuronal cell death and microRNA-323 upregulation
In this study, we performed the reoxygenation post-OGD (OGD/reoxygenation) experiment to produce ischemia/reperfusion in rat hippocampal neurons. The results shown during 6-24 h reoxygenation post-OGD, neuronal cell apoptosis was increased and cell survival was decreased (Figure 1A-C). Previous report showed that miR-323 was upregulated in rat spinal cord after ischemia/reperfusion treatment [19]. Consistently in our study, we detected the expression levels of miR-323 was upregulated in hippocampal neurons which subjected to OGD/reoxygenation. As shown in Figure 1D, the expression level of miR-323 was significantly decreased in neurons after OGD and continued to be downregulated during 6-24 h reoxygenation post-OGD when compared with the normoxic group.
Figure 1.
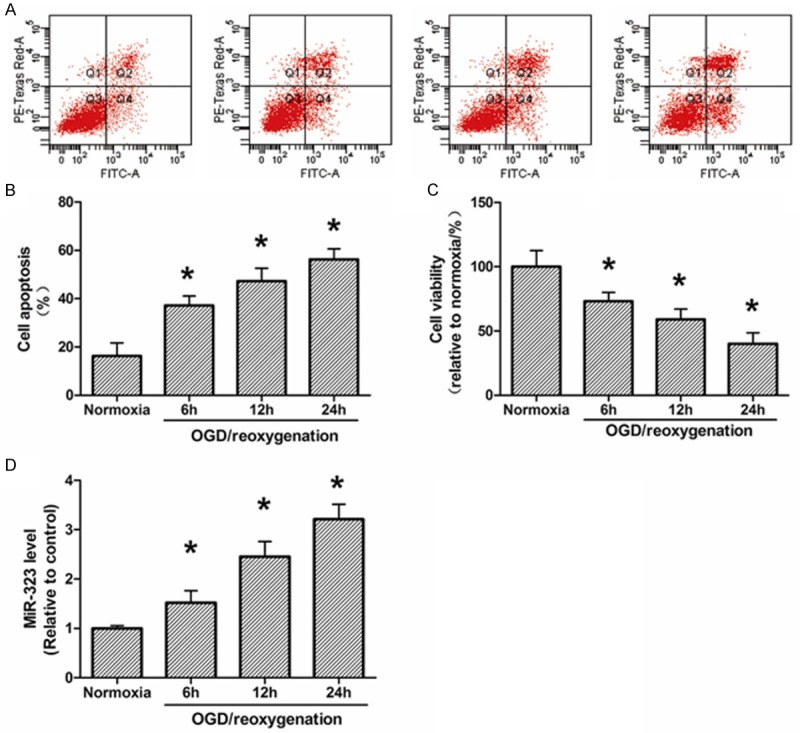
Apoptosis, survival and expression of miR-323 in hippocampal neurons after OGD-induced ischemic injury. A. Flow cytometry analysis of neuronal apoptosis during 6-24 h of reoxygenation post-3 h OGD. B. Quantization of A. C. XTT analysis of neuronal survival during 6-24 h of reoxygenation post-3 h OGD. D. The expression of miR-323 in neurons during 6-24 h of reoxygenation post-3 h OGD. The data presented as mean ± SD. *P<0.05 compared to the sham group. N=3, *P<0.05, compared with Normoxia.
miR-323 is involved in regulating OGD-induced neuronal cell death
To demonstrate the contribution of miR-323 in OGD induced ischemic injury, neurons were transfected with miR-323 mimics and anti-miR-323. As shown in Figure 2A, miR-323 expression levels were significantly upregulated or downregulated in hippocampal neurons after transfected with miR-323 mimics or anti-miR-323, respectively. Furthermore, we found that miR-323 upregulation accelerated the OGD/reoxygenation-induced cell apoptosis, where the miR-323 down regulation effectively inhibited OGD/reoxygenation-induced cell apoptosis as compared with that of blank or normoxia groups (Figure 2B). In contrast, miR-323 up regulation attenuated neuron survival under OGD/reoxygenation treatment, where the miR-323 downregulation effectively improved neuron survival under OGD/reoxygenation treatment as compared with that of blank or normoxia groups (Figure 2C).
Figure 2.
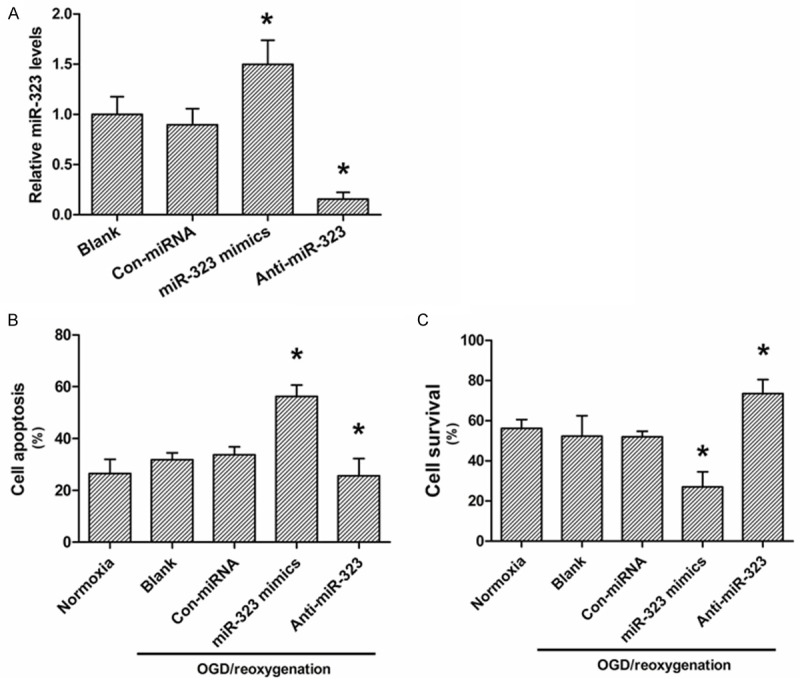
Effects of miR-323 expression on neuronal apoptosis after OGD-induced ischemic injury. A. Real-time PCR analysis of the miR-323 expression in neurons after miR-323 mimics or miR-323 inhibitor transfection. B. Flow cytometry analysis of neuronal apoptosis after miR-323 mimics or miR-323 inhibitor transfection under OGD-treatment condition. C. XTT analysis of neuronal survival after miR-323 mimics or miR-323 inhibitor transfection under OGD-treatment condition. The data presented as mean ± SD. *P<0.05 compared to the blank group.
MiR-323 directly modulates BRI3 expression in neuronal cell
The Target Scan database (http://www.targetscan.org) predicted that there is one binding site for miR-323 in the 3’UTR of BRI3 (positions 41-47, Figure 3A). A luciferase reporter assay was performed to analyze the interactions of miR-323 and BRI3. We employed vectors encoding a partial sequence of the 3’UTR of BRI3 mRNA where the predicted miR-323 target sites were located. We found that the luciferase activity was significantly reduced by co-transfection with miR-323 mimics and the vector carrying the 3’UTR of BRI3 compared with the negative control. On the contrary, co-transfection with the anti-miR-332 and the vector carrying the 3’UTR of BRI3 obviously increased the luciferase activity. These results showed that miR-323 bound directly to a specific site in the 3’UTR of BRI3 mRNA (Figure 3B). To confirm the role of miR-323 in the regulation of BRI3 expression, qRT-PCR and Western blot analyses were also performed. The qRT-PCR analysis showed that the transfection with miR-323 mimics or anti-miR-323 had no significant effect on the mRNA levels of miR-323 compared to the negative control (Figure 3C). The results of Western blotting showed that miR-323 mimics transfection significantly suppressed the protein levels of miR-323, whereas anti-miR-323 transfection dramatically promoted the protein levels of BRI3 compared to the negative control (Figure 3D). These data suggest that miR-323 suppressed the expression of BRI3 in a post-transcriptional manner.
Figure 3.

The effect of miR-323 on regulation of BRI3 expression in rat hippocampal neurons. A. Sequence alignment between miR-323 and the 3’-UTR of BRI3. B. Luciferase report assay was performed after co-transfection with BRI3 and miR-323 mimics or miR-323 inhibitor. C. The mRNA levels of BRI3 were determined after miR-323 mimics or miR-323 inhibitor transfection by real-time PCR. D. The protein levels of BRI3 were determined after miR-323 mimics or miR-323 inhibitor transfection by Western blot. E. Quantization of D. The data presented as mean ± SD. *P<0.05 compared to the negative control group.
BRI3 knockdown inhibited neuronal cell death and improved neuronal cell survival
Finally, we investigated the effect of BRI3 knockdown on neuronal cell apoptosis and survival. Rat neuronal cells treated with BRI3 siRNA showed significant blocking of the protein and mRNA levels of BRI3 compared to the control siRNA and control groups (Figure 4A, 4C). BRI3 siRNA treatment significantly inhibited neuronal cell apoptosis and improved neuronal cell survival compared to the control and control siRNA groups (Figure 4D, 4E).
Figure 4.
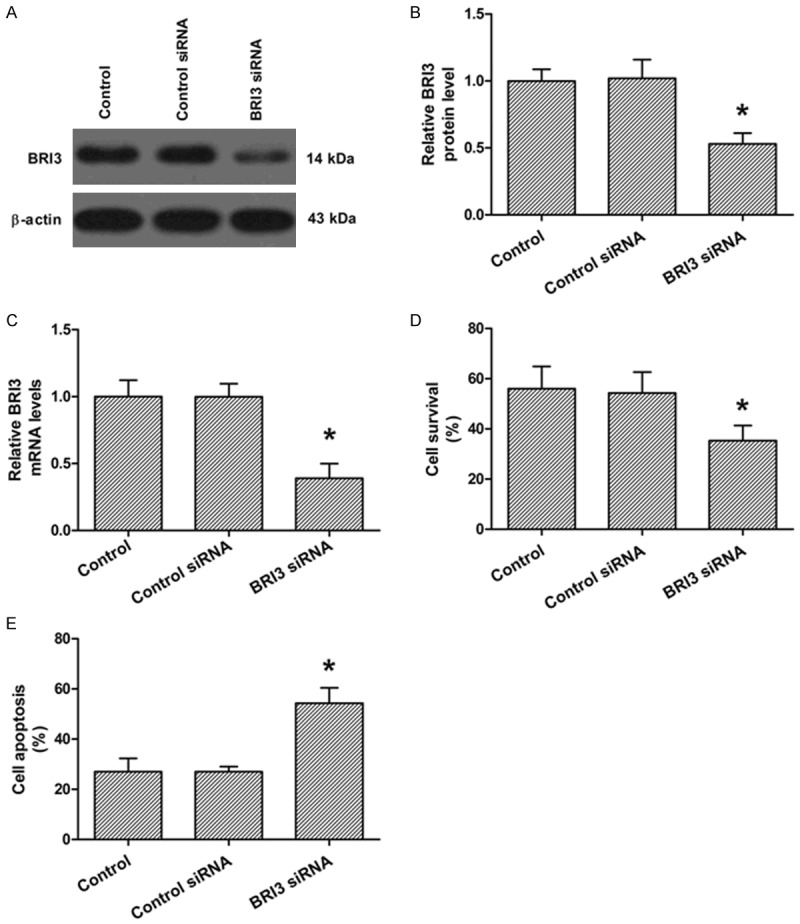
Effects of BRI3 expression on rat hippocampal neurons. A. Western blot analysis of the protein levels of BRI3 afterBRI3 siRNA or control siRNA transfection. B. Quantization of A. C. Real-time PCR analysis of the mRNA levels of BRI3 was determined afterBRI3 siRNA or control siRNA transfection. D. XTT analysis of neuronal survival after BRI3 siRNA or control siRNA transfection. E. Flow cytometry analysis of neuronal apoptosis after BRI3 siRNA or control siRNA transfection. The data presented as mean ± SD. *P<0.05 compared to the control group.
Discussion
Previous report discovered that miR-323 was up-regulated (more than 7-fold higher than normal rat) in the injured rat spinal cords at 48 h after I/R injury, but down-regulated in the atorvastatin pretreatment group, and atorvastatin plays protective effect on I/R-induced injury [19]. This phenomenon indicated that miR-323 is an important factor in I/R-induced injury. However, the probably role and precise mechanism of miR-323 in ischemic stroke or I/R-induced injury remains unclear.
In this study, we produced an OGD/reoxygenation model of cell ischemia in vitro. We found that OGD/reoxygenation treatment-induced apoptosis in rat hippocampal neurons. Consistently, we investigated that miR-323 was up-regulated after OGD/reoxygenation-treatment with a time-dependent manner. Moreover, up-regulation of miR-323 accelerated cell apoptosis induced by OGD/reoxygenation, whereas knockdown of miR-323 provided protective effects against ischemic injury.
Accumulation researchers provide evidence that miRNAs are involved in regulating I/R-induced neuronal injury by targeting a specific protein. For example, Wang et al. investigated that down-regulation of miRNA-30a alleviates ischemic injury through enhancing beclin 1-mediated autophagy [22]. Huang et al. suggests that downregulation of miR-134 alleviates ischemic injury through enhancing CREB expression and downstream genes [20]. MiR-124 improves I/R-induced neuronal death by targeting Ku70, and Ku70 plays a role in the regulation of ischemia/reperfusion (I/R)-induced apoptosis [23].
Brain protein I 3 (BRI3) was originally cloned as a type II transmembrane protein of 267 amino acids [24]. Northern blot analysis showed that BRI3 mRNA is expressed principally in the brain [25]. It has been reported that BRI3 can competitively inhibit amyloid precursor protein (APP) processing and may provide a new approach to AD therapy and prevention [26]. GUO et al. demonstrated that nifedipine treatment decreased miRNA-524-5p, resulting in the up-regulation of BRI3, and thereby leading to the proliferation and migration of breast cancer cells. Silencing BRI3 reversed the promoting effect of nifedipine on the breast cancer [27]. In the present study, we used target scan predictor and luciferase reporter assay confirmed that miR-323 directly targeted the 3’-UTR of BRI3 mRNA and that inhibition of miR-323 enhanced the expression of BRI3. Further experiment demonstrated that BRI3 knockdown suppressed neuronal survival and promote neuronal apoptosis under I/R treatment condition.
In summary, the present research demonstrated that I/R-induced neuronal apoptosis in vitro. Moreover, miR-323 plays an important role in the regulation of I/R-induced neuronal apoptosis via targeting BRI3. This study suggested that miR-323 might be a bright target for the prevention and therapy of cerebral ischemic injury. Additional experiments needed to perform to investigate the exact role of miR-323 in vivo.
Disclosure of conflict of interest
None.
References
- 1.Castillo X, Rosafio K, Wyss MT, Drandarov K, Buck A, Pellerin L, Weber B, Hirt L. A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J Cereb Blood Flow Metab. 2015;35:1561–9. doi: 10.1038/jcbfm.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan NC, Venketasubramanian N, Saw SM, Tjia HTL. Hyperhomocyst(e)inemia and Risk of Ischemic Stroke Among Young Asian Adults. Stroke. 2002;33:1956–1962. doi: 10.1161/01.str.0000021899.08659.c8. [DOI] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. Myocardial Reperfusion Injury. New Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 4.Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Rosenberg GA. Blood-Brain Barrier Breakdown in Acute and Chronic Cerebrovascular Disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo J, Cheng C, Chen CS, Xing X, Xu G, Feng J, Qin X. Overexpression of Fibulin-5 Attenuates Ischemia/Reperfusion Injury After Middle Cerebral Artery Occlusion in Rats. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9222-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH Oxidase Plays a Central Role in Blood-Brain Barrier Damage in Experimental Stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 8.Kamada H, Yu F, Nito C, Chan PH. Influence of Hyperglycemia on Oxidative Stress and Matrix Metalloproteinase-9 Activation After Focal Cerebral Ischemia/Reperfusion in Rats: Relation to Blood-Brain Barrier Dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min PK, Chan SY. The Biology of Circulating MicroRNAs in Cardiovascular Disease. Eur J Clin Invest. 2015;45:860–74. doi: 10.1111/eci.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell Joshua T, Olson Eric N. MicroRNAs in Stress Signaling and Human Disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 12.Eulalio A, Huntzinger E, Izaurralde E. Getting to the Root of miRNA-Mediated Gene Silencing. Cell. 2008;132:9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 13.Song X, Wang X, Arai S, Kurokawa R. Promoter-Associated Noncoding RNA from the CCND1 Promoter. Methods Mol Biol. 2012;809:609–622. doi: 10.1007/978-1-61779-376-9_39. [DOI] [PubMed] [Google Scholar]
- 14.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 18.Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs Expression and Function in Cerebral Ischemia Reperfusion Injury. J Mol Neurosci. 2014;53:242–250. doi: 10.1007/s12031-014-0293-8. [DOI] [PubMed] [Google Scholar]
- 19.Hu JR, Lv GH, Yin BL. Altered MicroRNA Expression in the Ischemic-Reperfusion Spinal Cord With Atorvastatin Therapy. J Pharmacol Sci. 2013;121:343–346. doi: 10.1254/jphs.12235sc. [DOI] [PubMed] [Google Scholar]
- 20.Huang W, Liu X, Cao J, Meng F, Li M, Chen B, Zhang J. miR-134 regulates ischemia/reperfusion injury-induced neuronal cell death by regulating CREB signaling. J Mol Neurosci. 2015;55:821–829. doi: 10.1007/s12031-014-0434-0. [DOI] [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2003–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, Han S, Li S, Li J. Down-Regulation of miRNA-30a Alleviates Cerebral Ischemic Injury Through Enhancing Beclin 1-Mediated Autophagy. Neurochem Res. 2014;39:1279–1291. doi: 10.1007/s11064-014-1310-6. [DOI] [PubMed] [Google Scholar]
- 23.Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. MicroRNA-124 (miR-124) Regulates Ku70 Expression and is Correlated with Neuronal Death Induced by Ischemia/Reperfusion. J Mol Neurosci. 2014;52:148–155. doi: 10.1007/s12031-013-0155-9. [DOI] [PubMed] [Google Scholar]
- 24.Deleersnijder W, Hong G, Cortvrindt R, Poirier C, Tylzanowski P, Pittois K, Van Marck E, Merregaert J. Isolation of Markers for Chondro-osteogenic Differentiation Using cDNA Library Subtraction: Molecular Cloning and Characterization of a Gene Belonging to a Novel Multigene Family of Integral Membrane Proteins. J Biol Chem. 1996;271:19475–19482. doi: 10.1074/jbc.271.32.19475. [DOI] [PubMed] [Google Scholar]
- 25.Vidal R, Calero M, Revesz T, Plant G, Ghiso J, Frangione B. Sequence, genomic structure and tissue expression of Human BRI3, a member of the BRI gene family. Gene. 2001;266:95–102. doi: 10.1016/s0378-1119(01)00374-2. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda S, Matsuda Y, D’Adamio L. BRI3 Inhibits Amyloid Precursor Protein Processing in a Mechanistically Distinct Manner from Its Homologue Dementia Gene BRI2. J Biol Chem. 2009;284:15815–15825. doi: 10.1074/jbc.M109.006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo DQ, Zhang H, Tan SJ, Gu YC. Nifedipine Promotes the Proliferation and Migration of Breast Cancer Cells. PLoS One. 2014;9:e113649. doi: 10.1371/journal.pone.0113649. [DOI] [PMC free article] [PubMed] [Google Scholar]