

Abstract
Sepsis induces hepatic injury but whether alpha-2 adrenoceptor (α2-AR) modulates the severity of sepsis-induced liver damage remains unclear. The present study used lipopolysaccharide (LPS) to induce hepatic injury and applied α2-AR agonist dexmedetomidine (DEX) and/or antagonist yohimbine to investigate the contribution of α2-AR in LPS-induced liver injury. Our results showed that LPS resulted in histological and functional abnormality of liver tissue (ALT and AST transaminases, lactate), higher mortality, an increase in proinflammatory cytokines (IL-1β, IL-6 & TNF-α), as well as a change in oxidative stress (MDA, SOD). Activation of α2-AR by dexmedetomidine (DEX) attenuated LPS-induced deleterious effects on the liver and block of α2-AR by yohimbine aggravated LPS-induced liver damage. Our data suggest that α2-AR plays an important role in sepsis-induced liver damage and activation of α2-AR with DEX could be a novel therapeutic avenue to protect the liver against sepsis-induced injury.
Keywords: Sepsis, α2-adrenoceptor agonist, dexmedetomidine, yohimbine, liver injury
Introduction
Despite recent advances in understanding the pathophysiology of sepsis, it still remains an enormous clinical challenge with an in-hospital mortality rate ranging from 17.9% to 27.8% in the US between 1975 and 2000 [1]. Between 1995 and 2005, the prevalence of severe sepsis in newborns more than doubled, from 4.5 to 9.7 cases per 1,000 births [2]. Sepsis was considered as a systemic inflammatory response to infection, and its severe form ultimately results in multiple organ failure. In sepsis, the liver is critical for host defense and tissue repair since it controls most of the coagulation and inflammatory processes. When this control is not adequate, a secondary hepatic dysfunction may occur, thereby resulting in multiple organ failure [3]. Cure of septic patients with liver damage complication is disappointingly with limited success, largely due to incomplete understating of the mechanisms underlying sepsis-induced liver injury. Standard septic shock therapy includes supportive treatment such as fluid resuscitation, administration of vasopressors, and respiratory and renal support [4]. Clinical trials of APC treatment reveal a relatively modest survival benefit in the selected septic patients [5]. Unfortunately, since it may lead to higher APACHE II scores, mortality, and major bleeding events [6,7], APC cannot be used in certain patients, such as infants, patients at risk for bleeding, and/or immunosuppressed patients, especially those with thrombocytopenia or neutropenia. Hence, other therapies are urgently needed to improve the survival rate in septic patients.
At present, sepsis is believed to be a neuro-immuno-endocrine disorder [8,9]. Complex network of cytokines, such as TNF-α, IL-1β, IL-6, inflammation-promoting transcription factor nuclear factor-κB (NF-κB) and HMGB1 have been implicated in the development of sepsis [8,9]. In a number of animal and human studies [10-13], α2-adrenoceptors (α2-AR) stimulation has been found to be beneficial for sepsis. Hsing et al found that dexmedetomidine (DEX), an α2 adrenoceptor (α2-AR) agonist, reduced sepsis-induced AKI by decreasing TNF-α and MCP-1 and increasing BMP-7 [13]. Arslan et al’s study indicated that DEX protected against lipid peroxidation and erythrocyte deformability alterations in experimental hepatic ischemia reperfusion injury [12]. In clinical studies, DEX infusion decreases TNF-α, IL-1, and IL-6 levels and IAP more than a propofol infusion [10]. Septic patients receiving DEX had more days free of brain dysfunction and mechanical ventilation and were less likely to die than those receiving a lorazepam-based sedation regimen [11]. Sezer et al reported that DEX exerted protective effects on liver histopathology during experimental sepsis in rats [14]. However, the role of α2-AR in sepsis-induced hepatic injury has not yet been explored. In light of the anti-inflammatory properties of α2-AR activation, we thus hypothesized that DEX, an α2-AR agonist, could mitigate sepsis-induced liver injury, and that yohimbine, an α2-AR antagonist, could aggravate liver damage induced by sepsis.
In the present study, we developed a rat model of sepsis by administrating bacterial endotoxin lipopolysaccharide (LPS) and evaluated liver damage after rats were co-treated with α2-AR agonist and/or antagonist. Our results reveal that DEX attenuated but yohimbine, a potent α2-adrenergic receptor antagonist, aggravated LSP-induced liver injury and suggest that stimulation of α2-AR with DEX could be beneficial to the septic patients with a complication of liver injury.
Materials and methods
Ethics statement
Animal experiment was performed in accordance with the protocol approved by the institutional animal care and use committees at Sun Yat-Sen University.
Animal model, experimental groups and survival rate determination
Male Sprague-Dawley rats weighing 200 to 300 g were housed in a temperature-controlled animal facility with alternating 12-hour light/dark cycles and fed standard laboratory chow ad libitum with free access to drinking water. Forty rats were randomly assigned to 5 groups (n = 8 in each group) using a random number generator (Excel 2007, Microsoft Office): 1) Control group: injected with an equivalent volume of 0.9% saline (i.v.) as other groups; 2) LPS-treated group: endotoxemia was induced by a bolus injection of Escherichia coli LPS derived from E. coli 0111:B4 (E. coli serotype 0111:B4-Sigma, injected intravenously at 15 mg/kg over 2 minutes); 3) LPS+DEX-treated group: dexmedetomidine was administered intravenously (i.v., 5 µg·kg-1·h-1) at 30mins after LPS administration (i.v., 15 mg/kg); 4) LPS+Yohimbine-treated group: injected with yohimbine (i.p. 250 μg/kg, Sigma-Aldrich) followed by LPS administration (i.v., 15 mg/kg) at 30 mins; 5) LPS+DEX+yohimbine-treated group: injected with yohimbine (i.p. 250 μg/kg) followed by LPS administration (i.v, 15 mg/kg) at 30 mins and DEX administration (i.v., 5 µg·kg-1·h-1) at 40 mins. The doses of LPS, DEX and yohimbine were based on previous organ protection studies [12,14-16].
Survival rate was determined over the time span of 6 hrs after LPS administration. All rats were sacrificed at 6 hrs following endotoxin infusion, and blood was collected from their abdominal aorta for blood plasma analysis. The samples were then centrifuged at 1200 g for 10 mins at 4°C, and the serum samples were stored at -80°C prior to assay. Rat livers were dissected and snap frozen in liquid nitrogen and stored at -80°C for further analysis.
Determination of serum levels of transaminases and lactate
Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT) concentrations were measured using TBA-200FR NEO from Toshiba Medical Systems Corp (Tochigi, Japan), according to the manufacturer’s instructions. Each sample was run in duplicate. Lactate was determined by using assay kits according to the manufacturer’s instructions (Pointe Scientific, Inc. Canton, MI).
Hematoxylin-eosin (HE) staining, TUNEL assay and detection of activated caspase-3
Liver tissue specimens were fixed in 10% neutral-buffered formalin and subsequently dehydrated through a graded ethanol series as described before [14]. After impregnation in paraffin wax, tissue samples were sectioned at 5 μm. Liver sections (4-5 μm) were stained with hematoxylin-eosin and examined by light microscopy using a Carl Zeiss microscope [17]. Liver cell apoptosis was assessed by using the terminal deoxynucleotidyltransferase-mediated nick end labeling-assay (TUNEL) staining using the TUNEL Apoptosis Assay Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions [17]. Each stained section was examined at high power fields (200×), and TUNNEL-positive cells in sinusoids were evaluated.
Determination of TNF-α, IL-6, IL-1β levels
The liver samples were excised, rinsed of blood, homogenized with polytron in an ice-cold lysis buffer (1% Triton X-100 in TBS with protease inhibitors, pH, 7.5) and sonicated for 30 seconds on ice. The tissue lysates were centrifuged at 12,000 rpm for 10 mins and protein concentration was measured by using Bio-Rad DC Protein Assay Kit (Bio-Rad, Hercules, CA). TNF-α, IL-1β and IL-6 in the liver and serum were quantified with the use of specific enzyme-linked immunosorbent assay (ELISA) kits according to the instructions provided by the manufacturer (BD Biosciences Pharmingen, San Diego, CA). Liver levels of TNF-α, IL-1β and IL-6 were normalized to the protein concentration in the sample.
Statistical analysis
All data are expressed as means ± SD (standard deviation) and compared by one-way analysis of variance (ANOVA) and Student-Newman-Keuls method. Unless otherwise stated, each group encompassed 8 animals (n = 8). Kaplan-Meier curves and log-rank test were used for survival analysis. P values of < 0.05 were considered as statistically significant.
Results
DEX ameliorated but yohimbine enhanced LPS-induced liver injury
It has been well-documented that LPS treatment induces hepatic dysfunction, as evidenced by an increase in liver-specific enzymes and lactate in the plasma. We set out to investigate whether DEX (a α2-AR agonist) could attenuate and yohimbine (a α2-AR antagonist) could aggravate LPS-induced increase in serum lactate and liver transanimases, such as alanine transaminase (ALT), aspartate transaminase (AST). As shown in Figure 1, serum levels of ALT, AST increased over 2-fold after LPS treatment (P < 0.01), suggesting a marked injury of liver tissue. DEX treatment decreased serum ALT and AST levels significantly by over 30%, while yohimbine considerably increased hepatic transaminases (P < 0.01). In addition, LPS elevated plasma lactate, an effect that could be attenuated by DEX treatment and aggravated by yohimbine. Histological examination of liver revealed hepatocyte necrosis, cytoplasmic vacuolization of hepatocytes, sinusoidal dilation and ballooning of hepatocytes, indicative of severe liver damage (Figure 2A). Consistent with the transaminases results, DEX attenuated but yohimbine aggravated LPS-induced histological change of liver tissue. TUNEL staining showed that there was significant hepatocyte apoptosis following LPS treatment and that DEX and yohimbine exerted opposite effects on hepatocyte apoptosis (Figure 2B). We further examined the effects of DEX treatment on the survival rate of the septic rats. As revealed, LPS+DEX group survived significantly longer than LPS group or LPS+yohimbine group (Figure 2C). Collectively, these results indicate that α2-AR is involved in LPS-induced hepatic injury.
Figure 1.
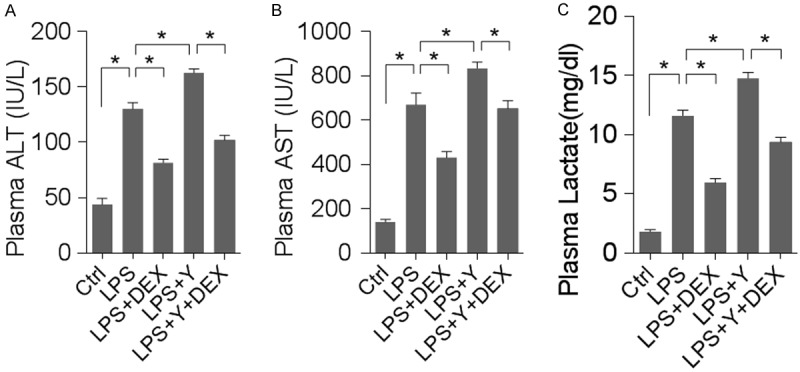
DEX attenuated but yohimbine aggravated LPS-induced liver injury (Ctrl = control; LPS = lipopolysaccharides; DEX = dexmedetomidine; Y = yohimbine). n = 8 animals for each group; *denotes P < 0.01. A: Plasma ALT levels in groups as indicated. B: Plasma AST levels. C: Lactate as measured in the blood plasma.
Figure 2.
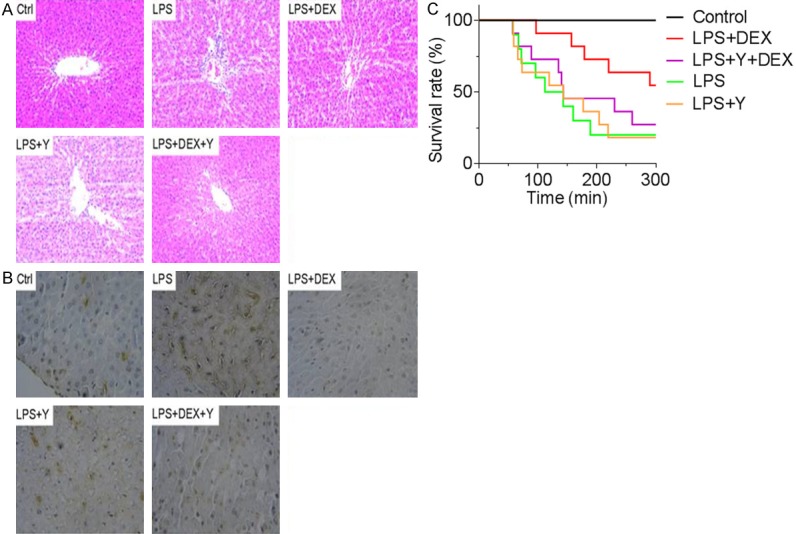
DEX attenuated but yohimbine aggravated histological change of liver tissue. A: HE staining of liver tissue in the groups as indicated. B: TUNEL staining of liver tissue. C: Animal survival rate in the indicated groups.
DEX lowered but yohimbine elevated LPS-induced increase in serum and liver proinflammatory cytokines
Given the histological and functional abnormality induced by LPS, it is plausible to hypothesize that inflammatory mediators contribute to liver injury. We therefore assayed 3 cytokines that have well-established roles in inflammation, i.e. IL-1β, IL-6 and TNF-α. As shown in Figure 3, LPS treatment elevated the levels of all 3 proinflammatory cytokines both in the plasma and in the liver tissue. Interestingly, activation of α2 AR by DEX significantly reduced the cytokine levels while block of α2 AR by yohimbine markedly increased cytokine concentrations. These data are in agreement with the histological analysis and the change in liver transanimases.
Figure 3.
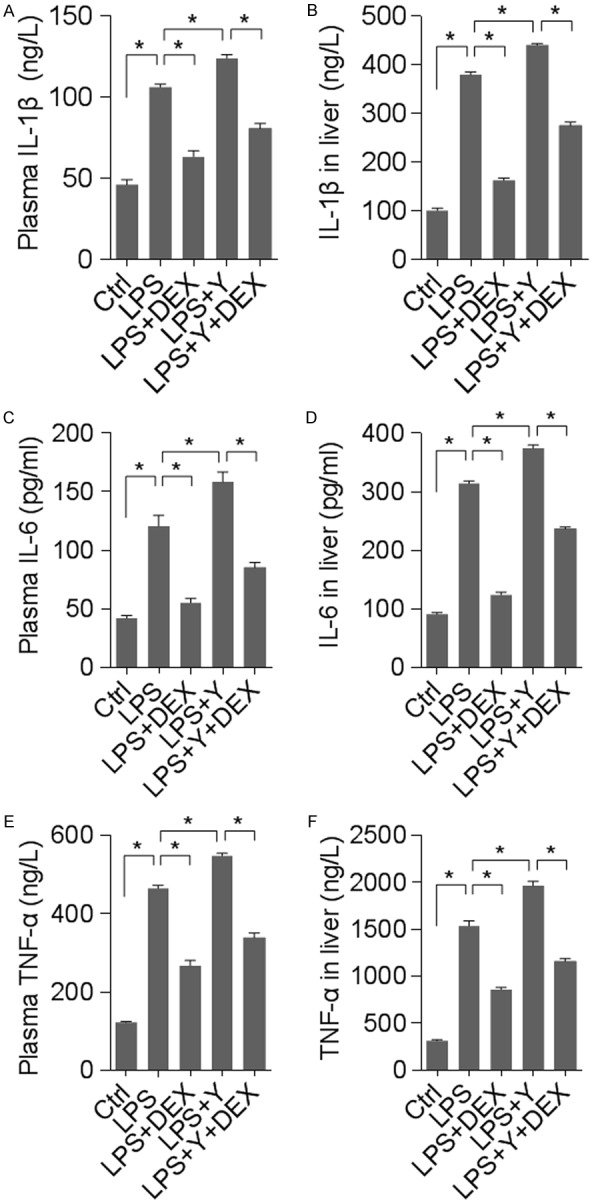
DEX lowered but yohimbine elevated LPS-induced rise in pro-inflammatory cytokines. n = 8 animals for each group; *denotes P < 0.01. (A, B) IL-1β levels in the blood plasma (A) and homogenated liver tissue (B). (C, D) IL-6 concentration in blood plasma (C) and liver tissue (D). (E, F) TNF-α level in blood (E) or liver tissue (F).
DEX treatment dampened but yohimbine instigated LPS-induced oxidative stress
In addition to proinflammatory cytokines, oxidative stress plays an important role in inflammation. Malondialdehyde (MDA) and superoxide dismutase (SOD) are often measured to represent the status of local or systemic oxidative stress. Measurement of MDA and SOD in the liver and plasma revealed that there was a marked rise in MDA and a significant decrease in SOD levels following LPS treatment; DEX-mediated α2-AR activation almost normalized MDA and SOD levels while yohimbine-mediated α2-AR blockade further enhanced LPS-induced alterations of MDA and SOD levels (Figure 4). These results suggest that α2-AR contributed to LPS-induced liver damage possibly by modulating oxidative stress.
Figure 4.
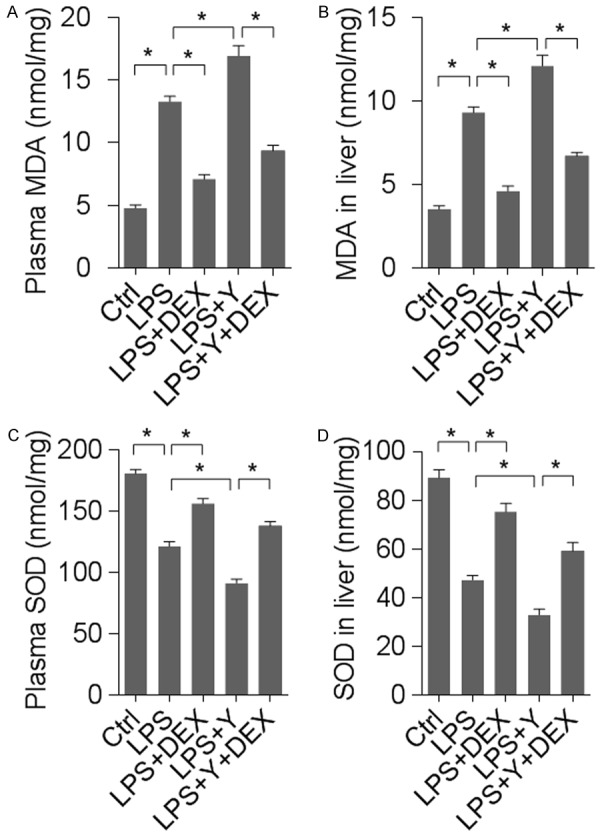
α2-AR activation or inhibition affected LPS-induced oxidative stress in liver. n = 8 animals for each group; *denotes P < 0.01. (A, B) MDA levels measured in the plasma (A) and the liver tissue (B). (C, D) SOD levels determined in the plasma (C) and the liver (D).
Discussion
Sepsis is one of the main causes of death in ICU patients [18,19]. Inflammation is the best studied mechanism responsible for the initiation of sepsis. A large number of cytokines, such as IL-6, IL-1β, TNF-α, are important inflammatory mediators in sepsis [4]. An imbalance in the production of anti- and pro-inflammatory cytokines provokes overwhelming inflammation and eventually leads to multiple organ failure [20,21]. In addition, marked oxidative stress as a result of sepsis-associated inflammatory responses initiates changes in mitochondrial function, thereby giving rise to hepatocyte apoptosis and organ damage [22].
The data we obtained showed that α2-AR stimulation with DEX significantly improved the histological structure and cellular function of the liver in LPS-induced sepsis, as evidenced by a decrease in liver transaminases and lactate; LPS-induced increase in pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, was lowered by α2-AR activation with DEX; DEX also normalized an LPS-induced imbalance in oxidative stress, as indicated by a decrease in MDA and an increase in SOD. All these beneficial effects of DEX were prevented by co-administration of an α2-AR antagonist yohimbine.
Sepsis causes microcirculation disturbance and tissue hypoxia, increasing lactate production. In the situation of hepatic dysfunction, an elevated serum lactate level may be due to either impaired lactate clearance or excessive production [23]. An elevated serum lactate level is associated with increased morbidity and mortality in patients with severe sepsis and septic shock [24]. In managing severe sepsis and septic shock, improved lactate clearance or suppressed lactate generation is an indicator of treatment success [25]. While the present study demonstrated that DEX treatment reduced plasma lactate levels and thereby improved microcirculation, future investigations might reveal whether this is due to enhanced lactate clearance or decreased production.
In sepsis, liver injury may be due to cytokines, oxidative stress and tissue hypoperfusion. In the present study, we assayed the cytokines and oxidative stress in both the plasma and the liver tissue and found that the results were nicely consistent. Considering the beneficial effects of DEX on the cytokine profile in the blood, it is conceivable that administration of DEX could protect the body from sepsis-induced injury of multiple organs. Indeed, it has been demonstrated that DEX is able to alleviate kidney, lung and brain injury in the context of sepsis or ischemia [13,15,26,27]. There are studies demonstrating that clonidine (centrally acting α2-AR agonists) treatment led to a reduction in leukocyte infiltration as well as their capacity to produce pro-inflammatory cytokines [28,29]. Although our results have established that DEX protects the liver in sepsis, it remains to be determined whether liver protection plays a causal role in the systemic attenuation of sepsis.
In vitro and some in vivo studies showed that local α2-AR stimulation provokes a pro-inflammatory response [16,30,31]. However, most animal studies and all human studies found the stimulation of local α2-AR to be anti-inflammatory. This dichotomous response may arise from different peripheral and central nervous system (CNS) actions of α2-AR agonists. Peripherally, α2-AR agonists may stimulate innate immunity. Centrally, the sympatholytic actions may enhance the relative parasympathetic tone to repress inflammation. Hence, peripheral α2-AR activation may evoke proinflammatory actions and central effects may shift towards an anti-inflammatory phenotype. There is evidence suggesting that the centrally acting α2-AR agonists such as clonidine, can reduce the central sympathetic tone by stimulating central α2-AR in the medulla oblongata [8,9]. Therefore, stimulation of central α2-AR reduces the tone of sympathetic nervous system and leads to a functional advantage in parasympathetic nervous system. Given that activation of parasympathetic nervous system is systemically anti-inflammatory, clonidine may reduce cytokines levels to avert septic shock without compromising the immune function to combat bacterial infection. Action of DEX on central α2-AR may partially account for its protection effects on sepsis and its associated complications, as DEX is more specific for central α2-AR compared with clonidine.
In summary, our data indicate that administration of DEX lessens the injury of liver caused by sepsis. Mechanistically, the beneficial effects of DEX in the treatment of sepsis-induced liver injury could be attributed to its several properties. DEX may dampen cytokine- and/or oxidative stress-mediated inflammation as well as mitigate cellular apoptosis, thus ameliorating sepsis.
Disclosure of conflict of interest
None.
References
- 1.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 2.Hartman ME, Linde-Zwirble WT, Angus DC, Watson RS. Trends in the Epidemiology of Pediatric Severe Sepsis. Pediatr Crit Care Med. 2013;14:686–693. doi: 10.1097/PCC.0b013e3182917fad. [DOI] [PubMed] [Google Scholar]
- 3.Dhainaut JF, Marin N, Mignon A, Vinsonneau C. Hepatic response to sepsis: interaction between coagulation and inflammatory processes. Crit Care Med. 2001;29:S42–47. doi: 10.1097/00003246-200107001-00016. [DOI] [PubMed] [Google Scholar]
- 4.Russell JA, Walley KR. Update in sepsis 2012. Am J Respir Crit Care Med. 2013;187:1303–1307. doi: 10.1164/rccm.201303-0567UP. [DOI] [PubMed] [Google Scholar]
- 5.Giamarellos-Bourboulis EJ. The failure of biologics in sepsis: where do we stand? Int J Antimicrob Agents. 2013;42(Suppl):S45–47. doi: 10.1016/j.ijantimicag.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Anger KE, Degrado JR, Greenwood BC, Cohen SA, Szumita PM. Evaluation of recombinant activated protein C for severe sepsis at a tertiary academic medical center. Ther Clin Risk Manag. 2013;9:277–284. doi: 10.2147/TCRM.S45412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fourrier F. Severe sepsis, coagulation, and fibrinolysis: dead end or one way? Crit Care Med. 2012;40:2704–2708. doi: 10.1097/CCM.0b013e318258ff30. [DOI] [PubMed] [Google Scholar]
- 8.MacLaren R. Immunosedation: a consideration for sepsis. Crit Care. 2009;13:191. doi: 10.1186/cc8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders RD, Hussell T, Maze M. Sedation & immunomodulation. Crit Care Clin. 2009;25:551–70. ix. doi: 10.1016/j.ccc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Tasdogan M, Memis D, Sut N, Yuksel M. Results of a pilot study on the effects of propofol and dexmedetomidine on inflammatory responses and intraabdominal pressure in severe sepsis. J Clin Anesth. 2009;21:394–400. doi: 10.1016/j.jclinane.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Pandharipande PP, Sanders RD, Girard TD, McGrane S, Thompson JL, Shintani AK, Herr DL, Maze M, Ely EW, investigators M. Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: an a priori-designed analysis of the MENDS randomized controlled trial. Crit Care. 2010;14:R38. doi: 10.1186/cc8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arslan M, Metin Comu F, Kucuk A, Ozturk L, Yaylak F. Dexmedetomidine protects against lipid peroxidation and erythrocyte deformability alterations in experimental hepatic ischemia reperfusion injury. Libyan J Med. 2012:7. doi: 10.3402/ljm.v7i0.18185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsing CH, Lin CF, So E, Sun DP, Chen TC, Li CF, Yeh CH. Alpha2-Adrenoceptor agonist dexmedetomidine protects septic acute kidney injury through increasing BMP-7 and inhibiting HDAC2 and HDAC5. Am J Physiol Renal Physiol. 2012;303:F1443–1453. doi: 10.1152/ajprenal.00143.2012. [DOI] [PubMed] [Google Scholar]
- 14.Sezer A, Memis D, Usta U, Sut N. The effect of dexmedetomidine on liver histopathology in a rat sepsis model: an experimental pilot study. Ulus Travma Acil Cerrahi Derg. 2010;16:108–112. [PubMed] [Google Scholar]
- 15.Ibacache M, Sanchez G, Pedrozo Z, Galvez F, Humeres C, Echevarria G, Duaso J, Hassi M, Garcia L, Diaz-Araya G, Lavandero S. Dexmedetomidine preconditioning activates pro-survival kinases and attenuates regional ischemia/reperfusion injury in rat heart. Biochim Biophys Acta. 2012;1822:537–45. doi: 10.1016/j.bbadis.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F, Wu R, Qiang X, Zhou M, Wang P. Antagonism of alpha2A-adrenoceptor: a novel approach to inhibit inflammatory responses in sepsis. J Mol Med (Berl) 2010;88:289–296. doi: 10.1007/s00109-009-0555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delladetsima I, Psichogiou M, Alexandrou P, Nikolopoulos G Revenas K, Hatzakis A, Boletis J. Apoptosis and hepatitis C virus infection in renal transplant recipients. Am J Clin Pathol. 2008;129:744–748. doi: 10.1309/U90671UBGT1GLKLL. [DOI] [PubMed] [Google Scholar]
- 18.Jaramillo-Bustamante JC, Marin-Agudelo A, Fernandez-Laverde M, Bareno-Silva J. Epidemiology of sepsis in pediatric intensive care units: first Colombian multicenter study. Pediatr Crit Care Med. 2012;13:501–508. doi: 10.1097/PCC.0b013e31823c980f. [DOI] [PubMed] [Google Scholar]
- 19.Moore LJ, Moore FA. Epidemiology of sepsis in surgical patients. Surg Clin North Am. 2012;92:1425–1443. doi: 10.1016/j.suc.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Spapen H. Liver perfusion in sepsis, septic shock, and multiorgan failure. Anat Rec (Hoboken) 2008;291:714–720. doi: 10.1002/ar.20646. [DOI] [PubMed] [Google Scholar]
- 21.Asfar P, De Backer D, Meier-Hellmann A, Radermacher P, Sakka SG. Clinical review: influence of vasoactive and other therapies on intestinal and hepatic circulations in patients with septic shock. Crit Care. 2004;8:170–179. doi: 10.1186/cc2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 23.Jansen TC, van Bommel J, Woodward R, Mulder PG, Bakker J. Association between blood lactate levels, Sequential Organ Failure Assessment subscores, and 28-day mortality during early and late intensive care unit stay: a retrospective observational study. Crit Care Med. 2009;37:2369–2374. doi: 10.1097/CCM.0b013e3181a0f919. [DOI] [PubMed] [Google Scholar]
- 24.Kang YR, Um SW, Koh WJ, Suh GY, Chung MP, Kim H, Kwon OJ, Jeon K. Initial lactate level and mortality in septic shock patients with hepatic dysfunction. Anaesth Intensive Care. 2011;39:862–867. doi: 10.1177/0310057X1103900510. [DOI] [PubMed] [Google Scholar]
- 25.Walker CA, Griffith DM, Gray AJ, Datta D, Hay AW. Early lactate clearance in septic patients with elevated lactate levels admitted from the emergency department to intensive care: Time to aim higher? J Crit Care. 2013;28:832–837. doi: 10.1016/j.jcrc.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Yang CL, Tsai PS, Huang CJ. Effects of dexmedetomidine on regulating pulmonary inflammation in a rat model of ventilator-induced lung injury. Acta Anaesthesiol Taiwan. 2008;46:151–159. doi: 10.1016/S1875-4597(09)60002-3. [DOI] [PubMed] [Google Scholar]
- 27.Okada H, Kurita T, Mochizuki T, Morita K, Sato S. The cardioprotective effect of dexmedetomidine on global ischaemia in isolated rat hearts. Resuscitation. 2007;74:538–545. doi: 10.1016/j.resuscitation.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 28.Romero-Sandoval A, Eisenach JC. Clonidine reduces hypersensitivity and alters the balance of pro- and anti-inflammatory leukocytes after local injection at the site of inflammatory neuritis. Brain Behav Immun. 2007;21:569–580. doi: 10.1016/j.bbi.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavand’homme PM, Eisenach JC. Perioperative administration of the α2-adrenoceptor agonist clonidine at the site of nerve injury reduces the development of mechanical hypersensitivity and modulates local cytokine expression. Pain. 2003;105:247–254. doi: 10.1016/s0304-3959(03)00221-5. [DOI] [PubMed] [Google Scholar]
- 30.Yang S, Zhou M, Chaudry IH, Wang P. Norepinephrine-induced hepatocellular dysfunction in early sepsis is mediated by activation of alpha2-adrenoceptors. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1014–1021. doi: 10.1152/ajpgi.2001.281.4.G1014. [DOI] [PubMed] [Google Scholar]
- 31.Weatherby KE, Zwilling BS, Lafuse WP. Resistance of Macrophages to Mycobacterium avium Is Induced by 2-Adrenergic Stimulation. Infect Immun. 2003;71:22–29. doi: 10.1128/IAI.71.1.22-29.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]