

Abstract
Aims: To study the clinicopathologic features of Stewart-Treves syndrome (STS) in postmastectomy patients including the epidemiology, presentation, morphology, differentiation, pathogenesis and therapeutic options. Methods and results: Ten cases of STS in postmastectomy patients were retrospectively identified in our archives, and immunohistochemistry for CD34, CD31, D2-40, HHV-8, CK, EMA and Ki-67 was performed. All ten patients presented with lymphedema after mastectomy as the first sign. Physical examination revealed multiple raised, pinkish-red papulo-vesicular lesions or ulceration as the early evidence of tumor in the field where radiation therapy was introduced. Microscopic examination revealed infiltrative proliferation of vessels and the heteromorphic tumor cells expressed CD34, CD31 and D2-40. Despite the various treatment modalities, 5 patients died in an average of 19 months, 4 patients survived to the last follow-up (9-31 months), and 1 patient got lost. Conclusions: STS is a fatal complication of postmastectomy lymphedema. Patients with STS have very poor prognosis. The key to improve patient’s survival is the early diagnosis through a high alert of this disease by primary care physicians and comprehensive physical examination of patients with pertinent history and suspicious clinical presentations followed by prompt biopsy for definitive diagnosis.
Keywords: Angiosarcoma, breast cancer, radiation, extremity swelling
Introduction
Angiosarcoma is a rare malignant neoplasm arising from the endothelium of lymphatic/blood vessel channels. Although de novo lesions and lesions associated with irradiation-associated lymphangioma have been described, its arising appears to be most commonly associated with chronic lymphedema of extremities [1,2]. Lymphedema after mastectomy for breast cancer, first described by Stewart and Treves in 1948 and known as Stewart-Treves syndrome, is a relatively rare tumor with an incidence varying form 0.07-0.45% [3,4]. It usually occurs 10 years after locoregional treatment with surgery and radiotherapy, and typically presents with a blue to red or purple macular or papular lesion on the swollen ipsilateral upper extremity of the breast with radical mastectomy and irradiation. The diagnosis can be delayed because these early lesions may be dismissed easily as traumatic ecchymosis by patients and physicians. It is a highly malignant tumor with aggressive clinical behavior. Although the advances in surgical technique, imaging and chemotherapy have remarkably improved the survival and limb salvage of patients with other common types of sarcomas [5], the prognosis of this rare secondary malignancy remains grim. We here report 10 such cases encountered in our practice to illustrate the clinical presentation, disease progression, differential diagnosis, prognosis, and to draw appropriate attention to early detection of this deadly malignant tumor so that the patient’s survival may be improved.
Material and methods
A comprehensive search of the archive was performed in the Department of Pathology, Beijing Shijitan Hospital, Capital Medical University, and 10 cases were identified from January 1, 2008 to September 1, 2014 with a diagnosis of angiosarcoma of the upper extremities in patients with status post mastectomy and axillary lymph node dissection for breast carcinoma. Nine patients were managed in our hospital and 1 patient was consulted in our department but treated in other hospital. The primary diagnosis was confirmed in each case by 2 of the authors after reviewing of the pertinent pathologic materials. Patient’s clinical information was collected with particular attention to the presence of edema of the upper extremity, the early presentation of symptoms and signs of tumors. The study was approved by the institutional review board for research.
All pathologic samples were initially fixed in 10% phosphate-buffered, neutral formaldehyde solution, processed automatically by standard procedure, and embedded in paraffin. The tissue blocks were retrieved for serial sections that were stained with hematoxylin-eosin. Immunohistochemistry for CD34 (1:100, Dako, USA), CD31 (1:100, Zymed, USA), D2-40 (1:100, Dako, USA), HHV-8 (1:50, Abcam, UK), CK-pan (1:60, Zymed, USA), EMA (1:60, Zymed, USA) and Ki67 (1:75, Dako, USA) was performed per manufactory protocols using Ventana automatic immunohistochemistry stainer. The primary antibodies were replaced by PBS in negative controls. All Sections were counterstained with 3, 3’-diaminobenzidine tetra-hydrochloride (DAB).
Cases were considered positive for CD34, CD31, D2-40, CK-pan and EMA if membrane/intracytoplastic immunoreactions were present in the tumor cells. Immunoreactivity for Ki67 was expressed as the percentage of the tumor cells counted across three representative fields. Cases were scored positive for HHV-8 if nuclear reactivity was present.
Results
Clinical Information (Table 1)
Table 1.
Clinical information of the patients
| Cases | Age | Date of surgery | Modus operandi | Symptom after radiation | Diagnosis of lymphangiosarcoma | Following up |
|---|---|---|---|---|---|---|
| 1 | 59 | 1999 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after radiation in 1 year, 2 months before diagnosis of agiosarcoma, blue macular was on upper extremity skin | 2008-04 | Die in 19 months |
| 2 | 67 | 1989 | Radical excision of right breast cancer | Ipsolateral upper extremity swelling after radiation in 1/2 year, 4 months before diagnosis of angiosarcoma, red macular was on upper extremity skin then coalesce into a fungoid mass | 2008-05 | Die in 15 months |
| 3 | 78 | 1993 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after radiation in 1 year, 2 months before diagnosis, many purple masses were on skin | 2008-07 | Die in 12 months |
| 4 | 50 | 1997 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after radiation in 1 year, 4 months before diagnosis of angiosarcoma, blue popular lesion was on skin, then ulcerated | 2008-12 | Die in 26 months |
| 5 | 54 | 1998 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after operation, 11 years later, many bleeding points and ecchymosis were on skin | 2009-10 | lost |
| 6 | 52 | 2000 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling for about 10 years, 2 months before diagnosis, many purple ecchymosis were on skin with ulceration | 2011-11 | Die in 25 months |
| 7 | 43 | 1998 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after radiation in 1 year, 1 month before diagnosis, many blue tubercles were on skin | 2012-07 | alive |
| 8 | 62 | 1999 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after operation, 4 months before diagnosis, many green purple masses were on skin all over the extremity, then burst with bloody fluid | 2012-08 | alive |
| 9 | 64 | 2000 | Radical excision of right breast cancer | Ipsolateral upper arm swelling for nearly 10 years, extension to forearm gradually, 5 months before diagnosis, many green purple masses were on skin of upper arm | 2014-04 | alive |
| 10 | 57 | 2004 | Radical excision of left breast cancer | Ipsolateral upper extremity swelling after operation, many green purple masses were on skin of left arm | 2014-05 | alive |
All patients were female, ranged from 43-78 year-old (median age 58.6 years), and had prior mastectomy for breast cancer. Eight patient with tumors occurred on the left and two on the right. All accepted radiation therapy and developed chronic swelling of the ipsilateral upper extremity after mastectomy.
The diagnosis of angiosarcoma was made 8 to 18 years (average 12.5 years) after mastectomy, with the mean interval of 11.8 years between the development of lymphedema and the present of angiosarcoma. Tumors involved the arm and forearm in 8 patients, confined to forearm in 1 patient and extended from arm to chest wall in another.
All patients except one (#4 underwent subsequent forequarter amputation after puncture biopsy) were underwent wide excision of the tumor, and 8 patients were followed by adjuvant therapy, among which 4 received chemotherapy, 2 accepted chemotherapy and radiation, and 1 was managed by immunotherapy. Two patients (#5 and #7) didn’t receive any additional treatments after wide excision. At the last follow-up by the end of March 2015, 4 patients were still alive (followed up 9-31 months), 5 patients died in average survival of 19 months, and 1 patient was lost.
Gross findings
Excisional specimens were received in 9 of the 10 cases. Grossly, blue-red or purple well defined macular or papular lesions were on the skin, and the small hemorrhagic nodules were discrete some coalesced into a fungoid mass (Figure 1). One Case was a biopsy specimen with skin and subcuits.
Figure 1.
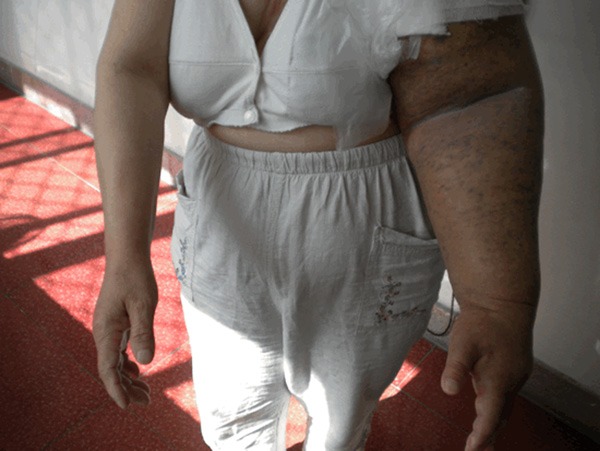
A patient’s clinical presentation: blue-red or purple well defined macular or papular lesions were on the skin, and the small hemorrhagic nodules were discrete, some coalesce into a fungoid mass.
Microscope findings
Pathological changes associated with chronic lymphedema were identified in all the cases: increased pigmentation of the basal layers and hyperkeratosis of the epidermis, mild lymphocytic infiltrates into the upper portion of the dermis, markedly thickened dermis, and varying degrees of edema in the interstitial tissues (Figure 2A). Striking lymphangiomatosis of the subcutaneous tissues was demonstrated, characterized by marked proliferation of predominant small lymphatic/vessel channels in the dermis lined by benign endothelial cells (Figure 2B). The diffuse scattering of many channels, however, lined by foci of large, hyperchromatic, proliferating endothelial cells frequently formed papillary intralumninal masse adjacent to the obvious malignant tumor tissue (Figure 2C, 2D). Besides such areas, the histological pattern varied widely among tumors, as well as among different areas of the same tumor. Well differentiated areas showed luminal pattern with numerous islands and cords of connective tissue surrounded by one to several layers of malignant cells. The cells were large, bizarre, and pleomorphic. The nuclei were gigantic and cytoplasm was scanty, and only occasional mitotic figure were present (Figure 2E). In contrast, in the undifferentiated areas, the tumor cells were arranged in a solid pattern (Figure 2F), consisting of large epithelioid cells with vesicular pleomorphic nuclei and multiple distinctive nucleoli. The cytoplasm was sparse, and slightly eosinophilic. Many mitosis were present (Figure 2G).
Figure 2.
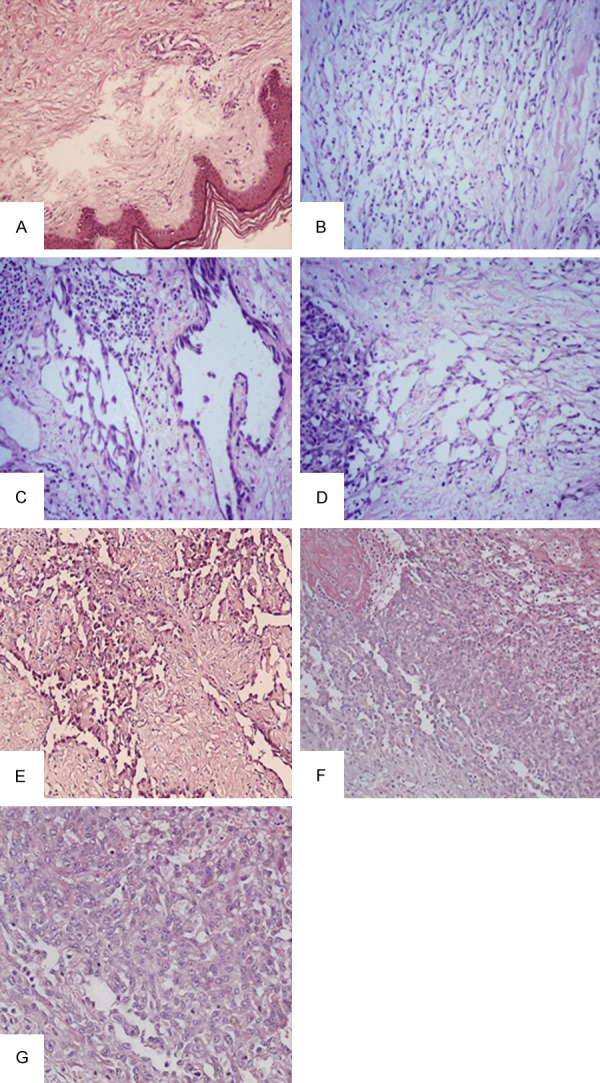
A: Hyperkeratosis of the epidermis, mild lymphocytic infiltrate in the upper part of the dermis, and varying degrees of edema in the interstitial tissues. (HE × 200); B: Marked lymphangiomatosis (HE × 200); C: Endothelial hyperplasia in the subcutaneous tissues (HE × 200); D: Marked lymphangiomatosis adjacent to the area of frankly malignant disease. (HE × 200); E: Well differentiated areas showed luminal pattern with numerous islands and cords of connective tissue surrounded by one to several layers of malignant cells. (HE × 200); F: Undifferentiated areas, the cells were arranged in a solid pattern. They were large, of epithelioid type, The cytoplasm was sparse, slightly eosinophilic. Between the cells was erythrocytes “lake”. (HE × 200); G: Many mitotic figures were seen. (HE × 400).
Immunohistochemical staining
Staining for CD34, CD31 (Figure 3A), D2-40 (Figure 3B, 3C) revealed a positive intracytoplastic and/or membrane reaction in the tumor cells focally or strongly. Immunohistochemistry for Ki67 was expressed as the percentage of tumor cells with nuclei staining by the total tumor cells counted across 3 representative fields which showed 30%-50% in our cases. There was no reaction for CK, EMA and HHV-8.
Figure 3.
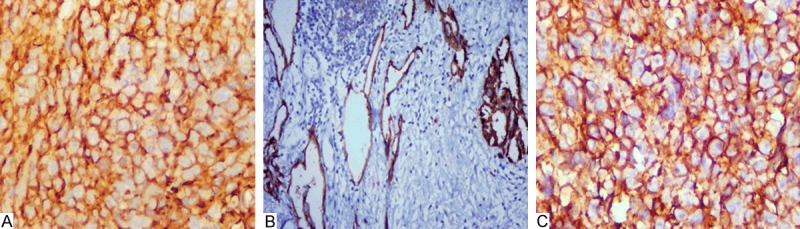
A: The tumor cells were stained by CD31 (× 400); B: D2-40 was positive on the membrane of the tumor cells. The left is lymphangiomatosis, the right is proliferating endothelial cells (× 200); C: D2-40 was positive on the membrane of the tumor cells (× 400).
Discussion
STS often presents with multifocal lesions, and usually appears after several years of lymphedema of the upper extremity, with a mean interval of 9-11 years and at a mean age of 61-63 years [6]. STS predominantly affects women with only 2 male cases of STS have been so far reported [7]. The classic clinical presentation is the appearance of blue-red or purple well defined macular or papular lesions on the arm, forearm, or hand of a patient. The lesions appear in small numbers, or as “crops”. In the later stages the lesions may coalesce and spread to chest wall and should girdle, and may eventually metastasize. In this small cohort of study, the patients were relatively older (43-78 years) and the tumors developed after a longer interval from the presence of lymphedema (mean 11.8 years). The possible reason to explain the difference may includes that (1) clinicians failed to recognize the disease at early stage and delay the time of biopsy; (2) some tumors were so well differentiated that the pathologists couldn’t make the correct diagnosis. So it is necessary to know this disease carefully and biopsy as early as possible.
The high incidence of severe, persistent lymphedema, the morbid lymphangiomatosis characterized by numerous newly formed lymphatic vessels with various stages of endothelial proliferation, and the multicentricity of the angiosarcoma, suggest that chronic lymphedema is the most significant risk factor for the development of STS. As the consequence of lymph node dissection, lymphedema induces lymphatic proliferation that probably forms the precondition to the eventual evolving into angiosarcoma [8,9]. Chronic lymphedema may also impair the immune environment in which the stimulus for the development of lymphatic collaterals leads to the onset of a neoplasm [10]. As a result, anatomic abnormality exists that interferes with the direct communication of the antigen and the regional lymph nodes, where immunocyte production normally occur [11]. Since an intact afferent lymph network and normal regional lymph nodes are important to the host ability to initiate immunity [11], it is conceivable that patients with chronic lymphedema have an impaired local immune responsiveness. Marek Stanczyk [12] et al illustrated pictures of a tumor center and edge by color lymphangiography. The lymphatic vessels on the tumor edge were characterized by the formation of an irregular network of bifurcated vessels and variable lumen diameter, whereas multiple lymphatic spaces and short, blind ending lymphatic vessels were seen in the tumor center. The findings suggest a defect in normal function. Taylor et al [13] have studied the protein content of edema fluid and found both qualitative and quantitative differences when compared to normal interstitial fluid. It is a possibility that the altered protein in an environment of lymph stasis alters the antigenic composition of the tissue. Edema fluid may also dilute the antigen from newly transformed neoplastic cells, thereby failing to promote a sufficient immune response.
Radiation therapy has been regarded as a pre-requirement for STS and radiation may serve as a contributing factor, perhaps by aggravating the lymphedema [14,15]. Valagussa et al [16] found that among 845 breast cancer patients who received radical or modified mastectomy alone without radiotherapy, no lymphedema was identified in any of their patients during 10 year’s follow-up. Huang and Maekillop [17] studied at cohorts of breast cancer patients with surgical excision, and found that the standardized incidence ratio (SIR) for STS was 3.8 in the group who received radiothehrapy (RDT), in comparing to a 1.4 SIR in the control group where RDT was not offered. A more dramatic increase in the development of angiosarcoma (AS), with a SIR of 26.2 was also noted in patients with RDT. On the other hand, the assertion has been challenged as many patients who developed STS had never received radiation [18].
Ongoing studies have reported non-random genetic alterations, including MYC amplification through gains in chromosome 8q24 in postradiation and lymphedema-associated AS of the breast and other organs [19]. The high predilection of MYC amplification in secondary AS over primary AS of the breast suggests a distinct pathogenic mechanism of AS in the setting of underlying lymphedema or prior irradiation [20].
Given the rarity of STS, clinicians should get familiar with the presentation and feature of this syndrome, particularly the lesions at early stage. High-risk patients are breast cancer patient who developed arm lymphedema after mastectomy, lymph node dissection, and radiation therapy. Clinicians should be prompt to excisional biopsy for any suspicious lesions in the upper extremity in postmastectomy patients, especially in the setting of chronic lymphedema. Pathological examination of the specimen is mandatory for a definitive diagnosis and immunohistochemistry can be confirmatory. Meanwhile, pathologists should always keep STS in mind and differentiate it from other mimics, which include lymphangioma, Kaposi sarcoma, adenocarcinoma and melanoma. Benign lymphangioma is composed of dilated lymphatic spaces, which are partially invested by a layer of smooth muscle and with occasional lymphoid aggregates. The tumor sometimes forms mutually traffic mesh. The lymphatic spaces contained either clear fluid or large numbers of foamy macrophages. The lymphatic endothelial cells lining the spaces were generally attenuated without any cytological atypism. Kaposi’s sarcoma is a proliferation of spindle endothelial cells with rare cytological presentation, forming thin vascular channels with red blood cell extravasation. In contrast, angiosarcoma is an atypical endothelial proliferation of epithelioid and fusiform cells, with common mitotic figures and necrosis. Angiosarcoma is often misdiagnosed as lymphangioma-like form of KS [21]. In addition to morphology, immunohistochemistry for HHV8 and molecular analysis can confirm the diagnosis [22,23]. Agiosarcoma may be composed of relatively solid sheets of anaplastic cells with many mitotic figures. This “medullary” pattern could easily be mistaken for highly undifferentiated metastatic adenocarcinoma of the breast. Immunohistochemical markers, such as CK and CD34, CD31, D2-40 could be very helpful for the distinction. The former is positive with CK, while the latter express CD34, CD31 and D2-40. Malignant melanoma cells are frequently epitheloid or spindle which may mimic angiosarcoma cells. However, melanoma cells may contain melanin pigments that vary in size or appear fine and “dusty” and the prominent nucleoli are often a feature, which are not seen in angiosarcoma. Their immunoprofiles are also different: the former is positive with S-100, HMB45, Melan-A, the latter is positive with CD34, CD31 and D2-40.
The therapy options are still limited for STS. Wide excision may be considered if regional control with clear margin can be accomplished. However, recurrence is always followed with wide excision alone [1,15,18]. Radical resection by forequarter amputation may provide the best tumor control, and early amputation appears to offer the best chance to prolong survival [1,24]. Even amputation may not guaranty free of local recurrence [25]. Patient #4 underwent a subsequent forequarter amputation after puncture biopsy, but unfortunately the patient died 26 months after diagnosis. Some investigators have proposed nonsurgical therapies before radical surgery, such as radiation, chemotherapy, and immunotherapy. Herrmann [26] recommended a thorough trial of both external and internal radiotherapy and before radical surgery should be used only if the tumor proves to be radioresistant. Chemotherapy is also used, and the protocol includes 5-fluorouracil, methotrexate, vincristine, actinomycin D, cyclophosphamide, doxorubin, dacarbazine, bleomycin, or any combination of these drugs. Chemotherapy may prolong survival if the initial response to the first treatment course is successful. Immunotherapy is a relatively new modality. Furue et al [27] reported a patient with STS who had severe dyspnea from a pleural effusion resulted from metastatic angiosarcoma to lung. Immunotherapy dramatically improved the patient’s dyspnea and pleural effusion. However, there is no survival benefits demonstrated yet.
STS patients have a very poor prognosis despite the various treatment modalities, with survival ranging from 19 to 31 months (mean survival of 20 months) [15,18,24]. Half of the patients die within 19 months of diagnosis [15]. In a literature review, Woodward [15] reported that only 11 of 121 (9%) patients survived longer than 5 years. STS patients are often plagued by local recurrence and most die of metastatic disease (usually to lung) within 2 years [4,28]. Five patients in our study died within 12 to 26 months after the initial diagnosis. The spread of angiosarcoma usually follows the subcutaneous veins to areas beyond the upper extremity. Metastases may occur to almost any organ hematogenously, especially the lung and bone. In our study, metastasis to lung and to bone was occurred in patient #4 and patients #1, 2, 3, 6 respectively.
At present, there is no ideal modality to treat STS. Preventive therapy seems to be the most promising approach at the present time. These include the prompt treatment of cellulitis to prevent further lymphatic blockage, and adequate management of lymphedema by pressure gradient therapy [29]. Of equal importance is the surgeon’s adherence to sound surgical principle and technique that avoids unnecessary interruption of lymph channels, especially around the axillary vein, during axillary lymph node dissection. The clinician should evaluate postmastectomy patients regularly and be prompt to biopsy of suspicious lesions. The key to improve survival of patients with STS is the early diagnosis through a high alert of this disease by primary care physicians and comprehensive physical examination of patients with pertinent history and suspicious clinical presentations followed by prompt biopsy for definitive diagnosis.
Disclosure of conflict of interest
None.
References
- 1.Taswell HF, Soule EH, Coentry MB. Lymphangiosarcoma arising in chronic lymphedematous extremities. Report of thirteen cases and review of literature. J Bone Joint Surg Am. 1962;44-A:277–294. [PubMed] [Google Scholar]
- 2.Necial M, Araújo de Pazos MÁ, de La Quintana Gordón M, Vázquez Rodríguez E. Malignant degeneration to lymphangiosarcoma of a chronic lymphedema in the lower left extremity. Cri Esp. 2015;93:e7. doi: 10.1016/j.ciresp.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: A report of six cases in elephantiasis lymphedema. Cancer. 1948;1:64–81. doi: 10.1002/1097-0142(194805)1:1<64::aid-cncr2820010105>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 4.Malhaire JP, Labat JP, Simon H, Le Maux H, Spindler P, Lucas B, Lamezec B. One case of Stewart-Treves syndrome successfully treated at two years by chemotherapy and radiation therapy in a 73-year-old woman. Acta Oncol. 1997;36:442–443. doi: 10.3109/02841869709001296. [DOI] [PubMed] [Google Scholar]
- 5.Sim FH, Pritchard DJ, Reiman HM, Edmonson JH, Schray MF. Soft-tissue sarcoma: Mayo Clinic experience. Semin Surg Oncol. 1988;4:38–44. doi: 10.1002/ssu.2980040109. [DOI] [PubMed] [Google Scholar]
- 6.Maddox JC, Evans HL. Angiosarcoma of the skin and soft tissue: a study of 44 cases. Cancer. 1981;44:1907–1921. doi: 10.1002/1097-0142(19811015)48:8<1907::aid-cncr2820480832>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 7.Oettle AG, van Blerk PJP. Postmastectomy lymphostatic endothelioma of Stewart and Treves in a male. Br J Surg. 1963;50:736–743. doi: 10.1002/bjs.18005022512. [DOI] [PubMed] [Google Scholar]
- 8.March WCH, Nurnberger F. The ultrastructure of Stewart-Treves syndrome. J Cutan Pathol. 1978;5:288–289. [Google Scholar]
- 9.Schreiber H, Barry FM, Russell WC, Macon WL 4th, Ponsky JL, Pories WJ. Stewart-Treves syndrome. A lethal complication of postmastectomy lymphedema and regional immune deficiency. Arch Surg. 1979;114:82–85. doi: 10.1001/archsurg.1979.01370250084018. [DOI] [PubMed] [Google Scholar]
- 10.Lasa MV, Mateo P, Bascón N, Baquedano J, Fuertes F, López P, Escó R. Lymphangiosarcoma in a chronic lymphedematous limb: a case report. Tumori. 1995;81:381–382. doi: 10.1177/030089169508100515. [DOI] [PubMed] [Google Scholar]
- 11.Fisher B. The present status of tumor immunology. Adv Surg. 1971;5:189–254. [PubMed] [Google Scholar]
- 12.Stanczyk M, Gewartowska M, Swierkowski M, Grala B, Maruszynski M. Stewart-Treves syndrome angiosarcoma expresses phenotypes of both blood and lymphatic capillaries. Chin Med J (Engl) 2013;126:231–237. [PubMed] [Google Scholar]
- 13.Taylor GW, Kinmonth JB, Dangerfield WG. Protein content of edema fluid in lymohedema. Br Med J. 1958;1:1159–1160. doi: 10.1136/bmj.1.5080.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brady MS, Garfein CF, Petrek JA, Brennan MF. Posttreatment sarcoma in breast cancer patients. Ann Surg Oncol. 1994;1:66–72. doi: 10.1007/BF02303543. [DOI] [PubMed] [Google Scholar]
- 15.Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremity. Cancer. 1972;30:562–572. doi: 10.1002/1097-0142(197208)30:2<562::aid-cncr2820300237>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 16.Valagussa P, Tancini G, Bonadonna G. Second malignancies after CMF for resectable breast cancer. J. Clin. Oncol. 1987;5:1138–1142. doi: 10.1200/JCO.1987.5.8.1138. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Maekillop AJ. Increased risk of soft tissue sarcoma after radiotherapy in women with breast carcinoma. Cancer. 2001;92:172–180. doi: 10.1002/1097-0142(20010701)92:1<172::aid-cncr1306>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 18.Sordillo PP, Chapman R, Hajdu SI, Magill GB, Golbey RB. Lymphangiosarcoma. Cancer. 1981;48:1674–9. doi: 10.1002/1097-0142(19811001)48:7<1674::aid-cncr2820480733>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Manner J, Radlwimmer B, Hohenberger P, Mössinger K, Küffer S, Sauer C, Belharazem D, Zettl A, Coindre JM, Hallermann C, Hartmann JT, Katenkamp D, Katenkamp K, Schöffski P, Sciot R, Wozniak A, Lichter P, Marx A, Ströbel P. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34–39. doi: 10.2353/ajpath.2010.090637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ginter PS, Mosquera JM, MacDonald TY, D’Alfonso TM, Rubin MA, Shin SJ. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709–716. doi: 10.1016/j.humpath.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez A, Laskin WB, Guitart J. Lymphangioma-like Kaposi sarcoma. J Cutan Pathol. 2005;32:286–292. doi: 10.1111/j.0303-6987.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- 22.Cheuk W, Wong KO, Wong CS, Dinkel JE, Ben-Dor D, Chan JK. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335–342. doi: 10.1309/B8TC-0LBV-H8XY-5MFV. [DOI] [PubMed] [Google Scholar]
- 23.Lasota J, Miettinen M. Absence of Kaposi’s sarcoma-associated virus (human herpesvirus-8) sequences in angiosarcoma. Virchows Arch. 1999;434:51–56. doi: 10.1007/s004280050304. [DOI] [PubMed] [Google Scholar]
- 24.Tomita K, Yokogawa A, Oda Y, Terahata S. Lymphangiosarcoma in postmastectomy lymphedema (Stewart-Treves syndrome): ultrastructural and immunohistologic characteristics. J Surg Oncol. 1988;38:275–282. doi: 10.1002/jso.2930380415. [DOI] [PubMed] [Google Scholar]
- 25.Noguchi M, Hasegawa H, Tajiri K, De Aretxabala X, Miyazaki I, Terahata S, Tomita K. Stewart-Treves syndrome. A report of two cases with a review of Japanese literature. Jpn J Surg. 1987;17:407–412. doi: 10.1007/BF02470642. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann JB. Lymphangiosarcoma of the chronically edematous extremity. Surg Gynecol Obstet. 1965;121:1107–1115. [PubMed] [Google Scholar]
- 27.Furue M, Yamada N, Takahashi T, Kikuchi K, Tsuchida T, Ishibashi Y, Kobori O, Ihara A, Kitayama J, Minami M. Immunotherapy for Stewart-Treves syndrome. Usefulness of ontrapleural administration of tumor-infiltrating lymphocytes against massive pleural effusion caused by metastastic angiosarcoma. J Am Acad Dermatol. 1994;30:899–903. [PubMed] [Google Scholar]
- 28.Miettinen M, Lehto VP, Virtanen I. Postmastectomy angiosarcoma (Stewart-Treves syndrome): light-microscopic, immunohistological, and ultrastructural characteristics of two cases. Am J Surg Pathol. 1983;7:329–339. [PubMed] [Google Scholar]
- 29.Chung KC, Kim HJ, Jeffers LL. Lymphangiosarcoma (Stewart-Treves syndrome) in postmastectomy patients. J Hand Surg Am. 2000;25:1163–1168. doi: 10.1053/jhsu.2000.18490. [DOI] [PubMed] [Google Scholar]