

Abstract
Tubulocystic renal cell carcinoma (TCRCC) is a rare, recently characterized RCC subtype with distinctive clinicopathologic and genetic characterizations as well as typical behaviors in an indolent fashion. However, sporadic case reports in the literature have indicated that TCRCC with sarcomatoid differentiation or poorly differentiated (PD) foci could behave aggressively. Herein, we reported two cases of TCRCC with PD foci indentified from our consultative service. Both patients were male and aged 66 y and 47 y, respectively. The first patient experienced radical nephrectomy while the other was treated by partial nephrectomy. Macroscopically, both tumors were described as partly cystic and solid with the greatest diameter measuring of 12-cm and 4.5-cm, respectively. Histologically, both lesions had classic areas of TCRCC occupying most part of the tumor with small papillary RCC component. In case one, PD foci were scatteredly distributed and mixed with TCRCC and papillary RCC components, while in the other case the PD foci were adjacent to the areas of TCRCC. In both tumors, the PD foci were composed of irregular, often angulated, small tubules lined by atypical eosinophilic cells and surrounded by desmoplastic stroma, resembling collecting duct carcinoma. Immunohistochemistry, in both tumors, both TCRCC component and PD foci showed the similar immunoprofiles, i.e., labeling strongly and diffusely with PAX8, AMACR and Vimentin, and focally with CK34βE12 but not with renal cell carcinoma marker or P63. In case one, the tumor invaded extensively into the adjacent renal parenchyma and focally into both renal sinusal and perirenal adipose tissues. The patient had metastasis in the pelvic cavity at the time of diagnosis and succumbed to the disease without further treatment 3 months later. The other case was organ confined but with focal positive renal parenchymal margin. The patient subsequently underwent radical nephrectomy and was in a good status without evidence of tumor recurrence or metastasis at a follow-up of 8 months.
Keywords: Tubulocystic renal cell carcinoma, tubulocystic carcinoma, RCC, collecting duct carcinoma, poorly differentiated foci
Introduction
Tubulocystic renal cell carcinoma (TCRCC) is a rare, recently characterized renal cell carcinoma (RCC) subtype and was not included in the latest edition of World Health Organization classification of renal tumors [1]. Tumors showing a similar morphology have been previously termed as “Bellinian epithelioma” or “low-grade collecting ductal carcinoma” in the literature [2,3]. It only received its current name in 2004 in a series of 29 cases presented in an abstract at the United State and Canadian Academy of Pathology meeting by Amin et al [3,4]. From then on, less than 100 TCRCC cases have been documented to date in the literature [5-9]. In 2012, TCRCC was recognized by the International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia [10] as one of the five new renal tumor entities due to its distinctive clinicopathologic and molecular genetic features. Histologically, TCRCC is characteristic of variable-sized tubules and cysts which are lined by a single layer of flat to hobnail cell with prominent nucleoli and separated by thin fibrous septa. TCRCC seems to have no relationship to collecting duct carcinoma based on the results of ultrastructural and gene expression analyses [7,9,11], and recent genetic and immunohistochemical studies have linked it with papillary RCC [6,8,12]. Biologically in its typical fashion, TCRCC behaved indolently with more than 90% affected patients having their tumor localized to the organ and those patients who have undergone entire tumor resection can survive for a long period of time without evidence of tumor recurrence or metastasis. However, approximate 10% patients could develop tumor recurrence or metastasis that are most frequent to bone, lymph node, live and pleura [6-9,13-15]. It has been shown that TCRCC occasionally demonstrated aggressive growth characteristics, such as poorly differentiated (PD) foci resembling collecting duct carcinoma [14,16], and sarcomatoid differentiation [17], increasing the risk of tumor recurrence and metastasis. In this study, we presented two cases of TCRCC with PD foci identified from our consultative service, and a literature review of its clinicopathologic features was also discussed.
Materials and methods
Two cases of TCRCC with PD foci were identified from our daily consultative service during 2011 to 2014. For both cases, all hematoxylin and eosin (H&E)-stained slides were available for review and one representative paraffin block was available for further analysis. Immunohistochemical studies using the avidin-biotin-complex immunoperoxidase technique were performed. The following commercially available antibodies were used: cytokeratin 7 (CK7), CK34βE12, PAX8, CD10, vimentin, renal cell carcinoma marker (RCCma), AMACR and P63. Appropriate positive and negative controls were run concurrently for all the markers tested. Macroscopic information was retrieved from the referring pathologic reports and follow-up information was obtained by clinical interviews.
Results
Case 1
The patient was a 66-year-old man who underwent laparoscopically radical nephrectomy for a huge right-kidney mass. Macroscopically, the tumor was described as partly solid and cystic, which was measured as 12-cm in greatest dimension and occupied most part of the kidney. Microscopically, the tumor was ill-circumscribed and invaded intensively into adjacent renal parenchyma and focally into both renal sinusal and perirenal fat. It consisted of three distinct histological components including classic tubulucystic RCC, type 2 papillary RCC and PD foci (Figure 1A-D), which were mixed with each other, occupying 70%, 20% and 10% of the entire tumor, respectively. Both TCRCC and papillary RCC components demonstrated their classic histological features (Figure 1A, 1B). While PD foci showed some appearances overlapping with collecting duct carcinoma, they were composed of irregular, often angulated, small tubules and glands with enlarged vesicular nuclei with prominent eosinophilic nucleoli identical to thoses seen in the tubulocystic and papillary components, setting in a inflammatory and desmoplastic stroma (Figure 1C, 1D). Mitotic figures were scant, necrosis and vascular tumor invasion were not present. Both the renal vein and Gerota’s fascia margins were negative, and adrenal gland showed no evidence of tumor invasion or metastasis.
Figure 1.
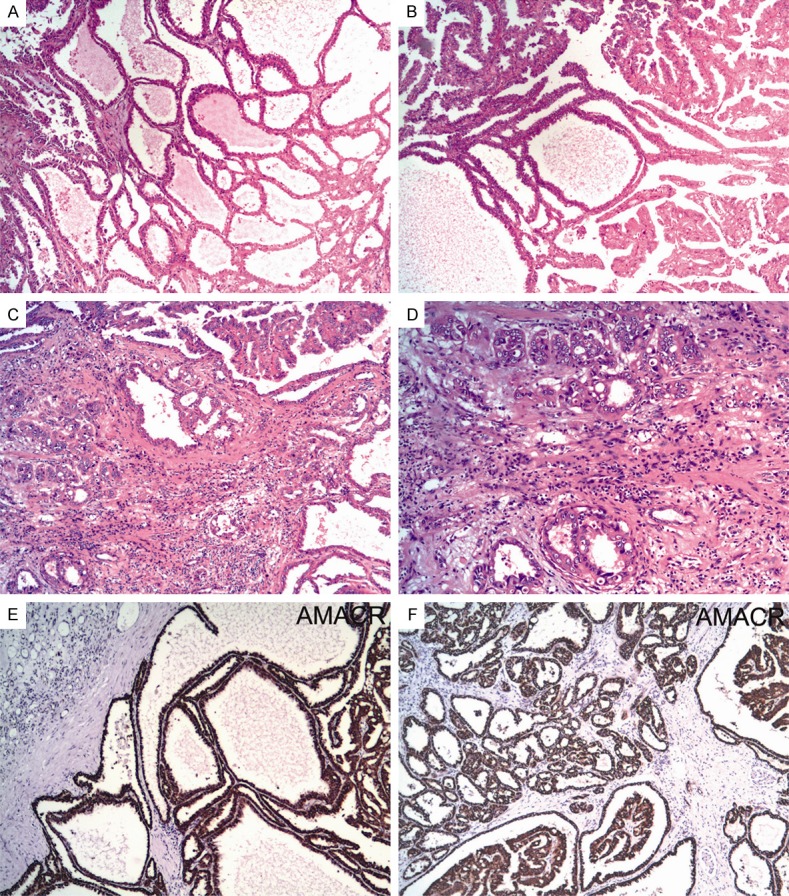
Histologically, case 1 had three distinct morphologic components including classic tubulucystic RCC (A), type 2 papillary RCC (B) and PD foci (C), which were mixed with each other. The PD foci were composed of irregular, angulated, small tubules and glands with enlarged vesicular nuclei and prominent eosinophilic nucleoli, setting in a inflammatory and desmoplastic stroma (D). Immunohistochemically, all the three components showed similar staining features with tumor cells labeling strongly and diffusely with AMACR (E, F).
Immunohistochemically, all of the three components showed similar staining features with tumor cells that were labeled strongly and diffusely with PAX8, AMACR (Figure 1E, 1F) and vimentin, focally with CK34βE12 but not with CK7, CD10, RCCma or P63. The patient had metastasis in the pelvic cavity at the time of diagnosis and succumbed to the disease without further treatment 3 months later.
Case 2
This patient was a 47-year-old male who underwent a partial nephrectomy. Macroscopically, the lesion was described as a cortical predominated, partly cystic and solid tumor with the greatest diameter measuring of 4.5-cm. Microscopically, the lesion had areas of typical TCRCC occupying 80% of the tumor (Figure 2A, 2B). Focally (less than 5%), a component of type 2 papillary RCC was observed. Proliferation of small tubules infiltrating adjacent to the areas of TCRCC with features resembling to those of collecting duct carcinoma was also identified. The PD carcinoma was measured as 1.3-cm and was composed of a mixture of poorly and more well-formed small tubules, nests, cords in a desmoplastic stroma (Figure 2C, 2D). Nuclei were markedly enlarged with prominent eosinophilic nucleoli. Mitotic figures were scant, necrosis and tumor vascular invasion were not noted. The renal parenchymal margin was focally involved by the tumor.
Figure 2.
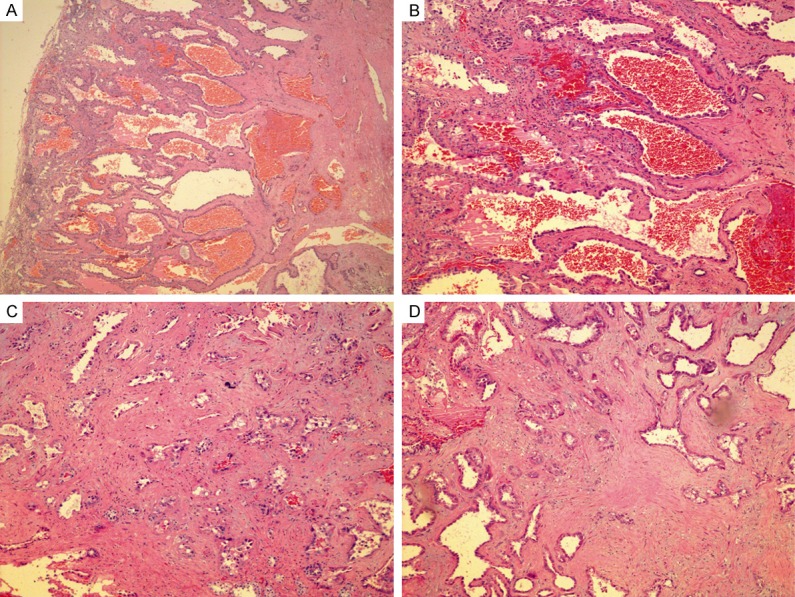
Microscopically, case 2 had areas of typical TCRCC occupying most part of the tumor (A, B), and foci of PD carcinoma (C, D) which were composed of a mixture of poorly and more well-formed small tubules, nests, cords in a desmoplastic stroma. Nuclei were markedly enlarged with prominent eosinophilic nucleoli.
Immunohistochemically, both TCRCC component and PD foci showed similar staining features with tumor cells that were labeled strongly and diffusely with PAX8 (Figure 3A, 3B), AMACR (Figure 3C, 3D), CD10 (Figure 3E, 3F) and vimentin, focally with CK7 and CK34βE12 but not with P63 or RCCma. The patient subsequently underwent radical nephrectomy and was in a good status without evidence of his tumor recurrence or metastasis at a follow-up of 8 months.
Figure 3.
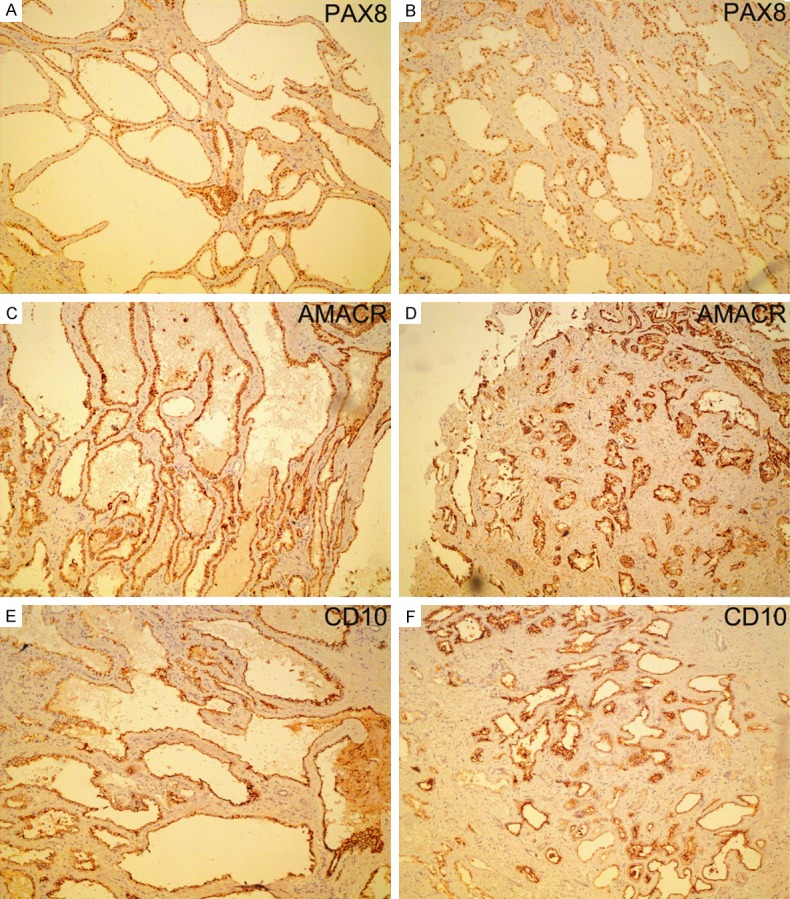
Immunohistochemically, both the TCRCC component (A, C, E) and PD foci (B, D, F) showed similar staining features with tumor cells labeling strongly and diffusely with PAX8, AMACR, and CD10.
Discussion
Tubulocystic RCC was first described as a unique morphotype of renal cell neoplasia in 1970s, being termed Bellinian epithelioma. Tumors showing a similar morphology were subsequently classified as low-grade collecting duct carcinoma [3,4]. More recently, several series have been reported and the name TCRCC has been applied. To date, less than 100 cases of these tumor have been documented, with tumors constituting to <1% of large series of RCC [5-9].
TCRCC has distinctive clinicopathologic and molecular genetic features. Clinically, it shows a strong male predisposition with male population affected seven-fold as frequently as female population with the mean age of 60 years at presentation (range: 15-94 years) [5-7,9,18]. Patients are usually asymptomatic, and on imaging, TCRCC is usually seen as a complex cyst, often type 3 or 4 in the Bosniak classification system [13]. The tumor mostly presents as a solitary mass and shows no relationship to any known syndromes, as also seen in our series. Macroscopically, TCRCC typically has a cortical or cortical-medullary epicenter and ranges in size from 0.5 to 17 cm in greatest dimension [6,7]. On cut surface, it is well-circumscribed, usually encapsulated, and has a characteristic “Swiss cheese” or “bubble wrap” appearance of white to gray color. Histologically, TCRCC is composed of well-formed, small to medium-sized tubules and cystically dilated larger tubules in varying proportions, which are separated by thin fibrous septa. The luminal spaces are lined by a single layer of atypical epithelial cells with abundant eosinophilic cytoplasm and often have, at least focally, a hobnail configuration. The nuclei are enlarged and have prominent nucleoli (ISUP grade 3). Mitoses are inconspicuous. The intervening stroma is generally thin, hypocellular and fibrotic, and occasional chronic inflammatory cells may be present. By immunohistochemistry, TCRCC expresses renal lineage differentiation transcription factor PAX2 or PAX8 [9], as also seen in our two cases, and shows protein expression of both proximal tubules and distal tubules/collecting ducts with tumor cells staining consistently positively for CK19, CD10, AMACR, and vimentin and less frequently for CK7, CK34βE12 and carbonic anhydrase IX (CA-IX) [5-9,11].
The histogenesis of TCRCC is unclear. Although considered initially to be of collecting duct origin, TCRCC has been shown to be distinct from collecting duct carcinoma based on the results of ultrastructural and gene expression analyses. Electron microscopic observations indicated that the tumor cells of TCRCC expressed aberrant tubular differentiation and had features of both proximal and distal nephron differentiation [7,9]. A comparative study looking at gene expression profiles of TCRCC and collecting duct carcinoma showed significant differences, indicating that these two tumor entities are unrelated [11]. Currently emerging evidence indicates that TCRCC is most closely related to papillary RCC, particularly type 2 papillary RCC [6,8,12,14]. Many series studies have documented that coexistence with TCRCC and papillary RCC in the same lesion was not just coincidence. In our series, both cases had small papillary RCC component similar to prior series. In addition to having morphological overlap with papillary RCC, TCRCC shares in many cases immunoprofiles and cytogenetic abnormalities. Both TCRCC and papillary RCC show diffuse and strong positive immunoreaction to AMACR, CD10, and vimentin and less frequently to CK7. Furthermore, gains in chromosomes 7 and 17 as well as loss of Y chromosome, which are characteristic genetic features of papillary RCC, are also commonplace in TCRCC as examined by fluorescence in situ hybridization (FISH) analysis [8,12,14].
With regard to differential diagnoses, TCRCC should be distinguished from cystic nephroma (CN), mixed epithelial and stromal tumor (MEST), oncocytoma, thyroid-like follicular RCC, acquired cystic disease-associated RCC (ACD-RCC), hereditary leiomyomatosis RCC syndrome-associated RCC (HLRCC-RCC), collecting duct carcinoma and renal medullary carcinoma. CN has a low nuclear grade and cellular stroma. MEST generally occurs in middle-aged women and contains ovarian-type stroma with or without smooth muscle differentiation [19]. Renal oncocytoma with tubulocystic pattern is composed of cells with deeply eosinophilic and granular cytoplasm and low nuclear grade. Additionally, an organoid pattern and edematous stroma have been reported [20]. In thyroid-like follicular RCC, glandular lumens contain eosinophilic, colloid-like materials often with absorbing vacuoles [21]. ACD-RCC occurs in end-stage kidneys and histologically may demonstrate a tubulocystic or tubulopapillay growth pattern with eosinophilic cells. However, cribriform/sieve-like architecture and intratumoral oxalate crystals, which are the most characteristic features of ACD-RCC [22], are absent in TCRCC. HLCRCC-RCC typically occurs in the setting of HLRCC with patients frequently harboring multiple cutaneous and uterine leiomyomas, and may have a morphology with tubulopapillary or cystic architecture resembling TCRCC. However, the distinctive nuclear feature of HLCRCC-RCC, a distinct prominent eosinophilic nucleolus with a clear halo similar to the cytology of a cytomegalovirus inclusion [23], is not present in TCRCC. Collecting duct carcinoma and renal medullary carcinoma typically occur in the renal medulla, and demonstrate a poorly differentiated adenocarcinoma, inflammatory infiltration, frequent perirenal fat invasion, lymphovascular invasion, intraluminal mucin and high nuclear grade [24]. By immunohistochemistry, both tumors express distal nephron differentiation markers but not proximal nephron differentiation markers such as AMACR, CD10, or vimentin, which are in contrast to TCRCC.
Despite its high grade cytology, most cases of TCRCC reported appear to have a favorable prognosis, usually being localized to the kidney at the time of diagnosis (pT1 and pT2) with <10% showing pT3 features [3,10]. Most tumors can be cured following surgery either by partial or radical nephrectomy; however, occasional tumors may develop recurrence and metastasis. Literature review of the biological behavior of all the reported TCRCC cases with follow-up revealed that, including one case of our series, 3 developed local recurrence [6,14,16] and 10 developed metastases to bone, live, peritoneum, pleura, lymph node, brain and plevic cavity [6-9,13-18]. TCRCCs showing aggressive growth features such as PD foci, as we and others [14] have reported, and sarcomatoid differentiation [17] seemed to behave more aggressively than those ordinary ones. In our series of 2 cases of TCRCC with PD foci, the PD areas of both consisted of collecting duct-like carcinoma areas with marked nuclear atypia and prominent nucleoli, occupying less than 15% of the tumor. One tumor invaded intensively into adjacent renal parenchyma and focally into both renal sinusal and perirenal fat, and the patient had metastasis in the pelvic cavity at the time of diagnosis and succumbed to the disease without further treatment 3 months later. The other one had focal positive margin followed by partial nephrectomy, and the patient subsequently underwent radical nephrectomy and was in a good status without evidence of tumor recurrence or metastasis at a follow-up of 8 months. In the initial report of a series of 3 cases of TCRCC with PD foci by Al-Hussain et al [14], 2 had PD foci occupying less than 20% of the tumor. In 2 cases, the PD areas consisted of collecting duct-like carcinoma areas, and 1 case had focal high-grade features with marked nuclear atypia and prominent nucleoli. Follow-up information available in two of their 3 patients showed that both tumors behaved aggressively with 1 patient dead of metastatic disease 9 months after surgery and the other one experiencing tumor recurrence 3 years postoperatively. Most recently, another single case [16] showing the same features of TCRCC with PD foci has also been reported where the patient developed two local recurrences and a brain metastasis during a follow-up of 6 years after the initial surgery. In addition, Bhullar et al [17] recently reported 1 case of TCRCC with tubulopapillary pattern and focal sarcomatoid areas that developed multiple peritoneal metastases, and the patient died 14 months after diagnosis. All the above mentioned evidence indicated that PD foci, when they occurring in the setting of TCRCC, although usually occupied only a minority of the tumor areas, can confer the tumor increasing risk of aggressive behavior. Sunitinib, a tyrosine kinase inhibitor which had been used to treat metastatic papillary and chromophobe RCC as well as urothelial carcinoma, has also resulted in a partial response for some patients with metastatic TCRCC [13,18,25]. However, the long term effect of this regimen awaits for further researches.
In summary, in the present study, we added another two cases of TCRCC with PD foci to the literature, and a detailed review of its histopathologic, immunohistochemical and prognostic features was also discussed. The fact that one of our patients had metastasis in the pelvic cavity at the time of diagnosis and eventually succumbed to the disease further confirmed the concept that TCRCC with PD foci increased the risk of aggressive behavior stronger than usual TCRCC. Surgical pathologists should pay more attentions to this tumor entity, whenever observed in the context of a classic TCRCC, the PD foci must be indicated in the pathological report.
Disclosure of conflict of interest
None.
References
- 1.Eble JN, Sauter G, Epstein JI, Sesterhenn IA. The World Health Organization Classification of Tumours of the Urinary System and Male Genital System. Lyon, France: IARC Press; 2004. [Google Scholar]
- 2.MacLennan GT, Farrow GM, Bostwick DG. Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology. 1997;50:679–684. doi: 10.1016/S0090-4295(97)00335-X. [DOI] [PubMed] [Google Scholar]
- 3.Delahunt B, Srigley JR. The evolving classification of renal cell neoplasia. Semin Diagn Pathol. 2015;32:90–102. doi: 10.1053/j.semdp.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Srigley JR, Delahunt B. Uncommon and recently described renal carcinomas. Mod Pathol. 2009;22(Suppl 2):S2–S23. doi: 10.1038/modpathol.2009.70. [DOI] [PubMed] [Google Scholar]
- 5.Azoulay S, Vieillefond A, Paraf F, Pasquier D, Cussenot O, Callard P, Sibony M. Tubulocystic carcinoma of the kidney: a new entity among renal tumors. Virchows Arch. 2007;451:905–909. doi: 10.1007/s00428-007-0483-7. [DOI] [PubMed] [Google Scholar]
- 6.Yang XJ, Zhou M, Hes O, Shen S, Li R, Lopez J, Shah RB, Yang Y, Chuang ST, Lin F, Tretiakova MM, Kort EJ, Teh BT. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. Am J Surg Pathol. 2008;32:177–187. doi: 10.1097/PAS.0b013e318150df1d. [DOI] [PubMed] [Google Scholar]
- 7.Amin MB, MacLennan GT, Gupta R, Grignon D, Paraf F, Vieillefond A, Paner GP, Stovsky M, Young AN, Srigley JR, Cheville JC. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol. 2009;33:384–392. doi: 10.1097/PAS.0b013e3181872d3f. [DOI] [PubMed] [Google Scholar]
- 8.Zhou M, Yang XJ, Lopez JI, Shah RB, Hes O, Shen SS, Li R, Yang Y, Lin F, Elson P, Sercia L, Magi-Galluzzi C, Tubbs R. Renal tubulocystic carcinoma is closely related to papillary re nal cell carcinoma: implications for pathologic classification. Am J Surg Pathol. 2009;33:1840–1849. doi: 10.1097/PAS.0b013e3181be22d1. [DOI] [PubMed] [Google Scholar]
- 9.Alexiev BA, Drachenberg CB. Tubulocystic carcinoma of the kidney: a histologic, immunohistochemical, and ultrastructural study. Virchows Arch. 2013;462:575–581. doi: 10.1007/s00428-013-1398-0. [DOI] [PubMed] [Google Scholar]
- 10.Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK, Zhou M, Argani P, Panel IRT. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol. 2013;37:1469–1489. doi: 10.1097/PAS.0b013e318299f2d1. [DOI] [PubMed] [Google Scholar]
- 11.Osunkoya AO, Young AN, Wang W, Netto GJ, Epstein JI. Comparison of gene expression profiles in tubulocystic carcinoma and collecting duct carcinoma of the kidney. Am J Surg Pathol. 2009;33:1103–1106. doi: 10.1097/PAS.0b013e3181a13e7b. [DOI] [PubMed] [Google Scholar]
- 12.Chen N, Nie L, Gong J, Chen X, Xu M, Chen M, Zhou Q. Gains of chromosomes 7 and 17 in tubulocystic carcinoma of kidney: two cases with fluorescence in situ hybridisation analysis. J Clin Pathol. 2014;67:1006–1009. doi: 10.1136/jclinpath-2014-202363. [DOI] [PubMed] [Google Scholar]
- 13.Hora M, Urge T, Eret V, Stransky P, Klecka J, Kreuzberg B, Ferda J, Hyrsl L, Breza J, Holeckova P, Mego M, Michal M, Petersson F, Hes O. Tubulocystic renal carcinoma: a clinical perspective. World J Urol. 2011;29:349–354. doi: 10.1007/s00345-010-0614-7. [DOI] [PubMed] [Google Scholar]
- 14.Al-Hussain TO, Cheng L, Zhang S, Epstein JI. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a series of 3 cases with fluorescence in situ hybridization analysis. Hum Pathol. 2013;44:1406–1411. doi: 10.1016/j.humpath.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 15.Iakovleva G, Iakovlev V, Ordon M, Srigley J, Yousef GM. Tubulocystic carcinoma of kidney: a challenging diagnostic entity mimicking multicystic kidney and presenting with bone metastasis. Histopathology. 2015;66:892–894. doi: 10.1111/his.12502. [DOI] [PubMed] [Google Scholar]
- 16.Sangle NA, Mao R, Shetty S, Schiffman JD, Dechet C, Layfield L, Agarwal N, Liu T. Novel molecular aberrations and pathologic findings in a tubulocystic variant of renal cell carcinoma. Indian J Pathol Microbiol. 2013;56:428–433. doi: 10.4103/0377-4929.125361. [DOI] [PubMed] [Google Scholar]
- 17.Bhullar JS, Thamboo T, Esuvaranathan K. Unique case of tubulocystic carcinoma of the kidney with sarcomatoid features: a new entity. Urology. 2011;78:1071–1072. doi: 10.1016/j.urology.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 18.Gizzi M, Aydin S, Machiels JP. Tubulocystic carcinoma of the kidney with fatal outcome in an adolescent male. Urol Int. 2015;94:485–487. doi: 10.1159/000366287. [DOI] [PubMed] [Google Scholar]
- 19.Moch H. Cystic renal tumors: new entities and novel concepts. Adv Anat Pathol. 2010;17:209–214. doi: 10.1097/PAP.0b013e3181d98c9d. [DOI] [PubMed] [Google Scholar]
- 20.Xiao GQ, Ko HB, Unger P. Telangiectatic oncocytoma: a previously undescribed variant of renal oncocytoma. Am J Clin Pathol. 2013;140:103–108. doi: 10.1309/AJCP9HDXYB2WYYJX. [DOI] [PubMed] [Google Scholar]
- 21.Amin MB, Gupta R, Ondrej H, McKenney JK, Michal M, Young AN, Paner GP, Junker K, Epstein JI. Primary thyroid-like follicular carcinoma of the kidney: report of 6 cases of a histologically distinctive adult renal epithelial neoplasm. Am J Surg Pathol. 2009;33:393–400. doi: 10.1097/PAS.0b013e31818cb8f5. [DOI] [PubMed] [Google Scholar]
- 22.Tickoo SK, dePeralta-Venturina MN, Harik LR, Worcester HD, Salama ME, Young AN, Moch H, Amin MB. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol. 2006;30:141–153. doi: 10.1097/01.pas.0000185382.80844.b1. [DOI] [PubMed] [Google Scholar]
- 23.Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–1585. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 24.Gupta R, Billis A, Shah RB, Moch H, Osunkoya AO, Jochum W, Hes O, Bacchi CE, de Castro MG, Hansel DE, Zhou M, Vankalakunti M, Salles PG, Cabrera RA, Gown AM, Amin MB. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol. 2012;36:1265–1278. doi: 10.1097/PAS.0b013e3182635954. [DOI] [PubMed] [Google Scholar]
- 25.Mego M, Sycova-Mila Z, Rejlekova K, Rychly B, Obertova J, Rajec J, Hes O, Mardiak J. Sunitinib in the treatment of tubulocystic carcinoma of the kidney. A case report. Ann Oncol. 2008;19:1655–1656. doi: 10.1093/annonc/mdn408. [DOI] [PubMed] [Google Scholar]