

Abstract
Objective: Aldosterone is related to the fibrosis of several organs, but the specific mechanism underlying the aldosterone induced hepatic fibrosis is still unclear. Methods: Separation, culture and identification of primary hepatic stellate cells (HSCs): The fluids and digestives used in the present study were able to completely remove blood cells, digest hepatocytes and matrix, and effectively separate HSCs. The in situ perfusion was performed at 2 steps: in situ perfusion with pre-perfusion fluid and ex vivo perfusion with enzyme-containing perfusion fluid. Influence of Ald on PAI-1 and Smad expressions in HSCs: cells were divided into control group, Ald group (10-6 M), spironolactone (SPI) group and Ald+SPI group, and the mRNA and protein expressions of PAI-1 and Smad were detected. Ald induced type I collagen expression in HSCs: Immunohistochemistry was performed to detect type I collagen expression in the supernatant of control group, Ald group (10-6 M), TGF-β1 group, and Ald+TGF-β1 group. Influence of Ald and TGF-β1 on PAI-1 expression in HSCs: cells were divided into control group, Ald group (10-6 M), TGF-β1 group, and Ald+TGF-β1 group, and the mRNA and protein expressions of PAI-1 were determined by RT-PCR and Western blot assay, respectively. Synergistic effect of Ald and TGF-β1 on PAI-1 expression in HSCs: cells were divided into control group, Ald group (10-6), TGF-β1 group, Ald (10-6 M)+TGF-β1 group, Ald (10-7 M)+TGF-β1 group and Ald (10-8 M)+TGF-β1 group, and the mRNA and protein expressions of PAI-1 were detected by RT-PCR and Western blot assay, respectively. Results: The survival rate, purity, markers and activation of HSCs were determined after separation. Influence of Ald on PAI-1 expression in HSCs: PAI-1 expression increased in HSCs of Ald group, SPI group and Ald+API group, and the PAI-1 expression in Ald group and Ald+SPI group was significantly higher than in control group (P<0.01). Influence of Ald on Smad expression in HSCs: Smad expression in Ald group, TGF-β1 group and ALD+TGF-β1 group was markedly higher than in control group (P<0.05). Smad expression in ALD+TGF-β1 group increased significantly when compared with Ald group (P<0.01). Ald induced type I collagen expression in HSCs: type I collagen expression in Ald group, TGF-β1 group and ALd+TGF-β1 group was dramatically higher than in control group (P<0.05), and it in ALd+TGF-β1 group was also significantly different from that in Ald group and TGF-β1 group (P<0.01). Synergistic effects of Ald and TGF-β1 on PAI expression in HSCs: PAI-1 expression in treated cells was markedly higher than in control group (P<0.01). PAI-1 expression in 10-6 M Ald+5 ng/ml TGF-β1 group increased dramatically as compared to Ald group and TGF-β1 group (P<0.01), but the increased PAI-1 expression reduced after SPI treatment. Ald at different concentrations exerts synergistic effect with TGF-β1 to increase PAI-1 expression in HSCs: PAI-1 expression in HSCs after different treatments increased markedly as compared to control group (P<0.01). Significant difference in PAI-1 expression was observed in 10-6 M Ald+50 pg/ml TGF-β1 group and 10-6 M Ald group (P<0.01), PAI-1 expression in 10-7 M Ald+50 pg/ml TGF-β1 group was significantly higher than in 50 pg/ml TGF-β1 group (P<0.01), but the PAI-1 expression in 10-7 M Ald+50 pg/ml TGF-β1 group was similar to that in 10-6 M Ald group (P>0.05). Conclusion: Aldosterone is able to activate HSCs and increase PAI-1 expression during hepatic fibrosis, which may be inhibited by spironolactone. Aldosterone and TGF-β1 may synergistically act on HSCs to increase PAI-1 expression as compared to treatment with aldosterone or TGF-β1 alone. Aldosterone or TGF-β1 alone may slightly increase PAI-1 expression in HSCs, which can be inhibited by spironolactone.
Keywords: Aldosterone, hepatic stellate cells, PAI-1, TGF-β1, spironolactone
Introduction
Aldosterone (Ald) is an important mineralocorticoid in humans and released after stimulation of angiotensin II, potassium and ACTH. Classically, Ald mainly acts on mineralocorticoid receptor (MR) to induce the water reabsorption and potassium excretion, which are crucial for the regulation of fluid and electrolytes. In addition, Ald has non-classic activities to function on epithelial tissues and non-epithelial tissues, inducing inflammation and organ fibrosis. Ald is one of important members of renin angiotensin-aldosterone system (RAAS), and its role in the liver diseases has been paid attention to. In hepatic cirrhosis, the RAAS is activated, and factors including angiotensin II (Ang II) may stimulate the synthesis and secretion of Ald, exerting multiple bioeffects. Ald is able to bind to and form a complex with MR on target cells, and this complex then translocates into nucleus to regulate the expression of some target genes including ENaC and Na+-K+-ATP-ase, modulte the metabolism of water and salt, and induce sodium and water retention, resulting in hypertension [1]. Moreover, Ald may also activate NADPH oxidase to increase reactive oxygen speeies (ROS), which promotes the inflammation [2,3]. Ald may up-regulate the expressions of type I and IV collagens, transforming growth factor [3,4], connective tissue growth factor (CTGF) [5,6] and plasminogen activator inhibitor-1 (PAI-1) [7], leading to the aggregation of extracellular matrix (ECM).
Hepatic fibrosis is a shared pathophysiology in chronic liver injury and also a consequence of liver injury. Hepatic fibrosis has been found to be one of causes of portal hypertension and liver dysfunction and also a risk factor of liver cancer. Hepatic stellate cells (HSCs) are major cells producing collagens during the liver injruy and have been found to be a key factor involved in the occurrence and development of fibrosis. Ald has been found to be a pivotal pathogenic factor in the fibrosis [8]. It is one of potent factors causing collagen synthesis and mitosis, and may promote the fibrosis of some organs including heart, lung and kidney [9]. In the presence of factors able to induce liver injury, the occurrence and development of fibrosis are as a result of interaction of these factors. However, little is known on the role of Ald as an independent stimulus in the hepatic fibrosis.
In recent years, studies on fibrotic diseases reveal that PAI-1 is a pro-fibrotic factor and plays important roles in the pathogenesis of fibrotic diseases. PAI-1 is an important member of fibrinolytic system and a major physiological inhibitor of tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). Under the pathological condition, high PAI-1 expression may inhibit the plasminogen activator which decreases the fibrin degradation and increases the ECM deposition. Increased PAI-1 expression has been found to be associated with renal tubular sclerosis and renal interstitial fibrosis [10-12]. In some studies, increasing attention has been paid to PAI because TGF-β1 as a crucial factor regulating fibrosis may induce PAI expression, and TGF-β1 over-expressed mice with glomerulosclerosis have concomitant high PAI-1 expression [13]. It has been confirmed that TGF-β1 may induce the PAI-1 synthesis, and there is a TGF-β1 response element at the promoter of PAI-1 gene [14]. Ald may increase TGF-β1 expression [15]. Available studies also demonstrate that Ald is able to increase the expression of pro-fibrotic factors, leading to pathological consequences. Ald may interact with Ang II to elevate PAI-1 expression, which promotes the deposition of ECM among vascular endothelial cells [16]. These findings have been found in Ald treated mesangial cells of rats [17]. In addition, Ald may also increase the PAI-1 mRNA expression in wide-type mice [18], and PAI-1 expression is closely associated with sclerosis in mice with radiation induced renal injury [12]. Finally, blood Ald concentration is related to PAI-1 expression [19], which further supports the regulatory role of Ald in PAI-1 expression. TGF-β1 is a potent pro-fibrotic factors and able to increase the synthesis and secretion of ECM and block the degradation of ECM. As a downstream gene of TGF-β1, PAI-1 also involves in the fibrosis of tissues and organs and might play a special role in the pathogenesis of fibrotic diseases. Studies on the role of PAI-1 in fibrotic diseases may provide new targets for the therapy of these diseases. Thus, the present study was undertaken to investigate the effect of Ald on the PAI-1 expression in hepatic stellate cells (HSCs) in vitro, which may provide evidence for the Ald induced hepatic fibrosis and identify therapeutic targets for hepatic fibrosis.
Materials and methods
Separation, culture and identification of primary HSCs
The liver of male SD rats was perfused with enzymes at 2 steps for the digestion, and HSCs were harvested after removal of confounding cells by density gradient centrifugation. HSCs were observed and counted under a microscope. The survival rate was determined after trypan blue staining. HSCs were identified by immunocytochemistry for desmin. After Ald treatment for 48 h, immunohistochemistry was performed to detect α-SMA expression in HSCs, aiming to evaluate the HSCs activation.
Detection of PAI-1 mRNA expression by RT-PCR
Monolayer cells were treated with TRIzol for lysis, followed by addition of chloroform and a 15-s shaking. The mixture was incubated at room temperature for 3 min and centrifuged at 12000× g for 15 min at 4°C. PCR amplification was performed using Taq polymerase (Katara) in a total volume of 50 μl. The upstream and downstream primers for rat PAI-1 mRNA were 5’-CCTCCTCATCCTGCCTAAGTT-3’ and 5’-CTTGACCTTTTGTAGTGCTTGTG-3’ RNA in water was precipitated with isopropanol at room temperature for 10 min, followed by centrifugation at 12000× g for 10 min at 4°C. The supernatant was removed, and RNA washed in 75% ethanol. Centrifugation was done at 12000× g for 5 min at 4°C. The supernatant was removed, and RNA air-dried for 5-10 min at room temperature. The RNA purity was determined, and PAI-1 mRNA expression was detected by RT-PCR.
Detection of PAI-1 and Smad protein expression by Western blot assay
Total protein was extracted from HSCs and subjected to SDS-PAGE. Proteins were transferred onto PVDF membrane which was then blocked in 5% non-fat mild and treated with primary antibody at 4°C over night. After incubation with secondary antibody at room temperature for 2 h, visualization was done with chemiluminescence method.
Detection of type I collagen expression by immunohistochemistry
Cells were seeded into 96-well plates at a density of 1×106 cells/L. Samples were diluted and added to plates. Deparaffinization and dehydration were done in xylene and ethanol at different concentrations, respectively. Antigen retrieval was performed in citrate buffer. Sections were then treated with 5% H2O2 to inactivate endogenous peroxidase. After blocking in goat serum for 20 min at 37°C, sections were incubated with primary antibody (1:100) at 4°C over night and then with biotin conjugated secondary antibody (1:100) at 37°C for 30 min. Visualization was done with DAB, and counterstaining with hematoxylin, followed by fixation.
Results
Survival rate, purity, marker and morphology of HSCs
Cells were counted as follow: cell count = (total cells in 4 big grids/4) ×104 × volume (mL), cell counting was done twice for each sample, and an average was obtained. Survival rate was determined after trypan blue staining. Results showed the survival rate of HSCs was 95.0±1.2% after separation. Under an inverted phase contrast microscope, the newly separated HSCs were round, had small cell volume and lipid droplets in the cytoplasm, and presented high refraction. After culture for 24 h, cells were adherent to the wall, began to spread, became oval and had reduced refraction. After culture for 48 h, a majority of cells were adherent and extended pseudopods. At 7 days, these cells enlarged, became stellate and irregular, and presented focal growth (Figure 1).
Figure 1.
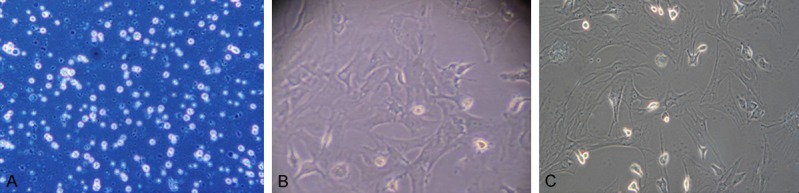
Survival rate, purity, marker and morphology of HSCs. A. HSCs after trypan blue staining (×10); B. HSCs after 3-day culture (×20); C. HSCs after 7-day culture (×20).
Identification of HSCs by immunohistochemistry
Immunocytochemistry was performed to detect desmin expression, aiming to identify HSCs. Positive cells had brown granules in the cytoplasm and their nuclei were blue. The cell purity was higher than 90% (Figure 2).
Figure 2.
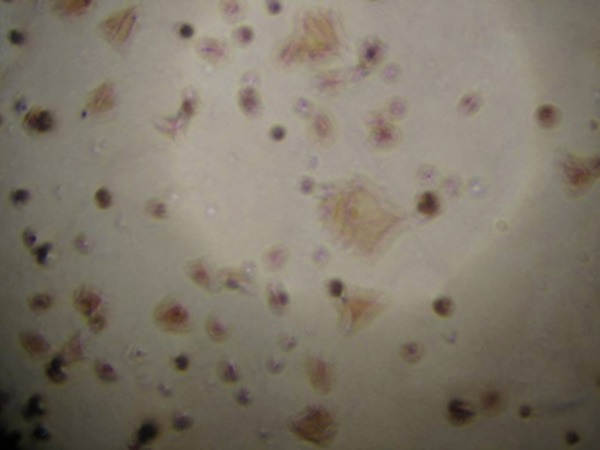
Immunocytochemistry for desmin (×10).
HSCs activation after Ald treatment
Immunohistochemistry was done to detect α-SMA expression in HSCs. Results showed α-SMA expression increased significantly in HSCs after Ald treatment for 24 h, and HSCs had filamentous structures (Figure 3).
Figure 3.

Immunohistochemistry for α-SMA in HSCs. A. (×40). B. (×10).
Effect of Ald and spironolactone (SPI) on the PAI-1 expression in HSCs
Cells were divided into control group, 10-6 M Ald group, 10-5M spironolactone (SPI) group and Ald+SPI group. Results showed PAI-1 expression in Ald group, SPI group and Ald+SPI group was markedly different from that in control group (P<0.01). In addition, significant differences were also observed in PAI-1 mRNA and protein expressions between Ald group and Ald+SPI group (P<0.05). (Figure 4).
Figure 4.
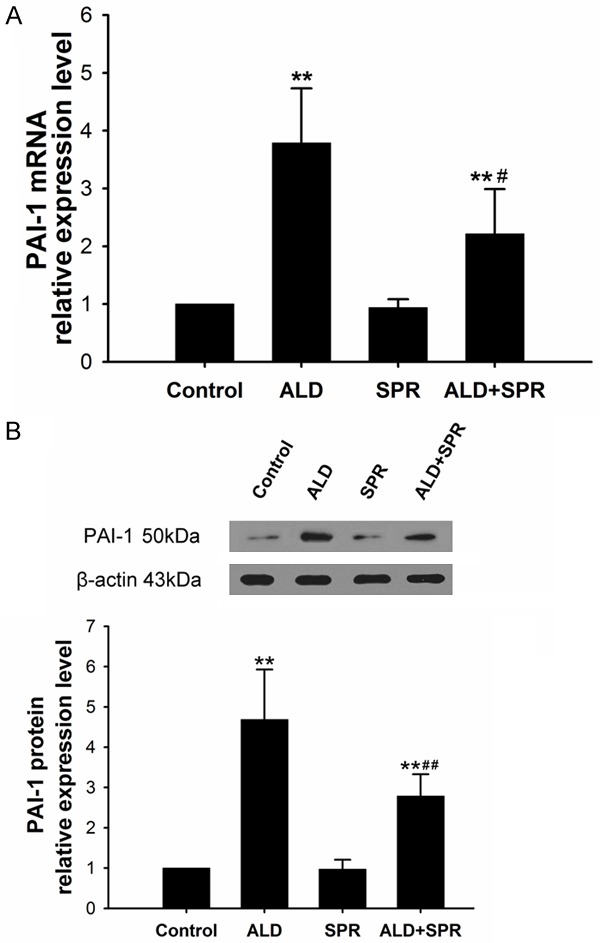
A. Effect of Ald on PAI-1 mRNA expression in HSCs. B. Effect of Ald on PAI-1 protein expression in HSCs. *vs control group; #Ald+SPI group vs Ald group; **P<0.01.
Effect of Ald on Smad expression in HSCs
The Smad expression in 10-6 M Ald group, 5 ng/ml TGF-β1 group and 10-6 M Ald+5 ng/ml TGF-β1 group was significantly higher than in control group (P<0.05), and marked difference was also observed between Ald+TGF-β1 group and Ald group (P<0.01) and between Ald+TGF-β1 group and TGF-β1 group (P<0.05) (Figure 5).
Figure 5.
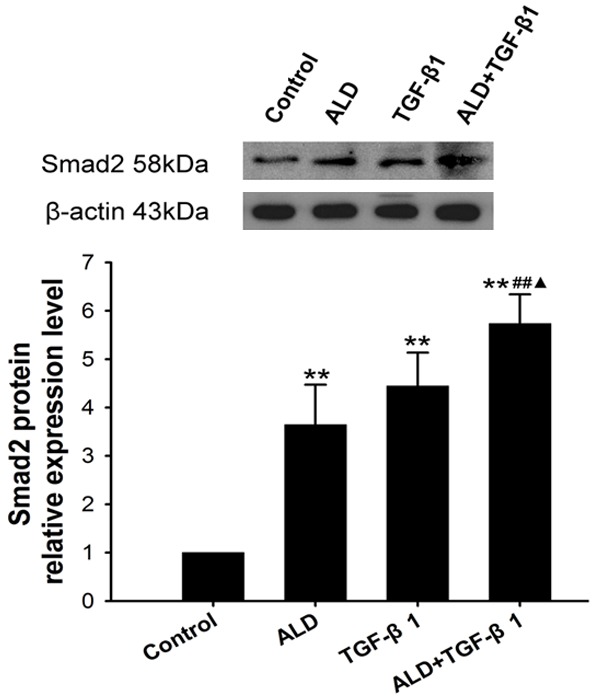
Effect of Ald on Smad protein expression in HSCs. *vs control group, #ALD+TGF-β1 group vs ALD group, ▲ALD+TGF-β1 group vs TGF-β1 group. *P<0.05, **P<0.01, ***P<0.001.
Effect of Ald on type I collagen expression in HSCs
The type I collagen expression in 10-6 M Ald group, 5 ng/ml TGF-β1 group and 10-6 M Ald+5 ng/ml TGF-β1 group was markedly higher than in control group (P<0.01), and type I collagen expression in Ald+TGF-β1 group increased significantly as compared to Ald group and TGF-β1 group (P<0.01) (Figure 6).
Figure 6.
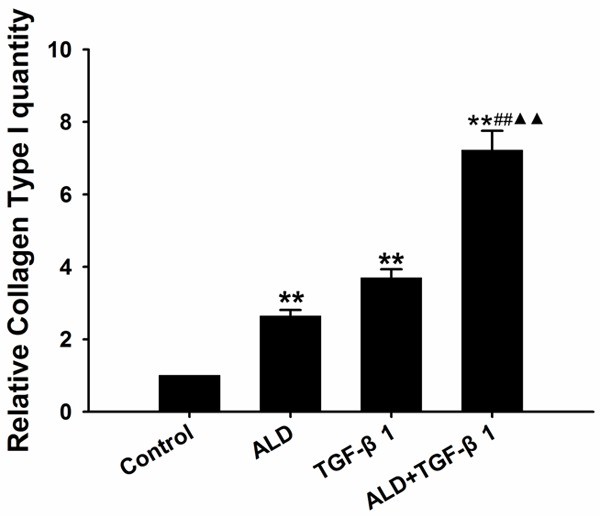
Ald induced type I collagen expression in HSCs. *vs control group, #ALD+TGF-β1 group vs ALD group, ▲ALD+TGF-β1 group vs TGF-β1 group. *P<0.05, **P<0.01, ***P<0.001.
Effect of Ald and TGF-β1 on PAI-1 expression in HSCs
PAI-1 expression in 10-6 M Ald group, 5 ng/ml TGF-β1 group, and 10-6 M Ald+5 ng/ml TGF-β1 group was markedly higher than in control group (P<0.01). Moreover, the PAI-1 expression in Ald+TGF-β1 group increased dramatically as compared to Ald group and TGF-β1 group (P<0.01) (Figure 7).
Figure 7.
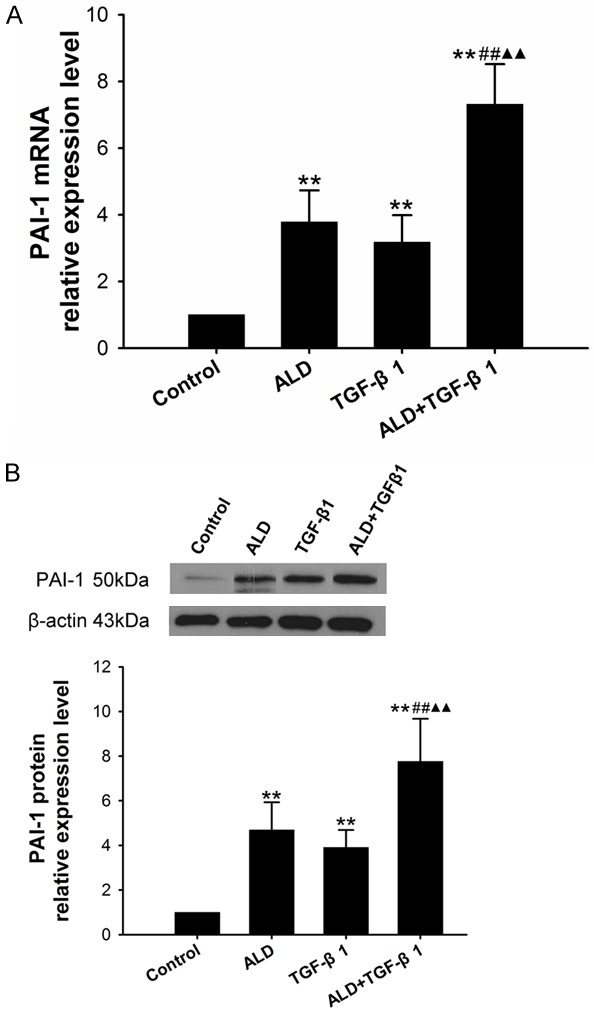
A. Effect of Ald and TGF-β1 on the PAI-1 mRNA expression in HSCs. B. Effect of Ald and TGF-β1 on the PAI-1 protein expression in HSCs. *vs control group, #10-6 M Ald+TGF-β1 group vs TGF-β1 group, ▲10-6 M Ald+TGF-β1 group vs ALD group. *P<0.05, **P<0.01, ***P<0.001.
Effect of Ald at different concentration and TGF-β1 on PAI expression in HSCs
PAI-1 expression in Ald group, 50 pg/ml TGF-β1 group, 10-6 M Ald+TGF-β1 group, 10-7 M Ald+TGF-β1 group and 10-8 M Ald+TGF-β1 group was significantly higher than that in control group (P<0.01). Moreover, significant difference was observed between 10-6 M Ald+TGF-β1 group and Ald group (P<0.01), and between 10-7 M Ald+TGF-β1 group and TGF-β1 group (P<0.01), but the PAI-1 expression was comparable between 10-7 M Ald+TGF-β1 group and Ald group (P>0.05) (Figure 8).
Figure 8.
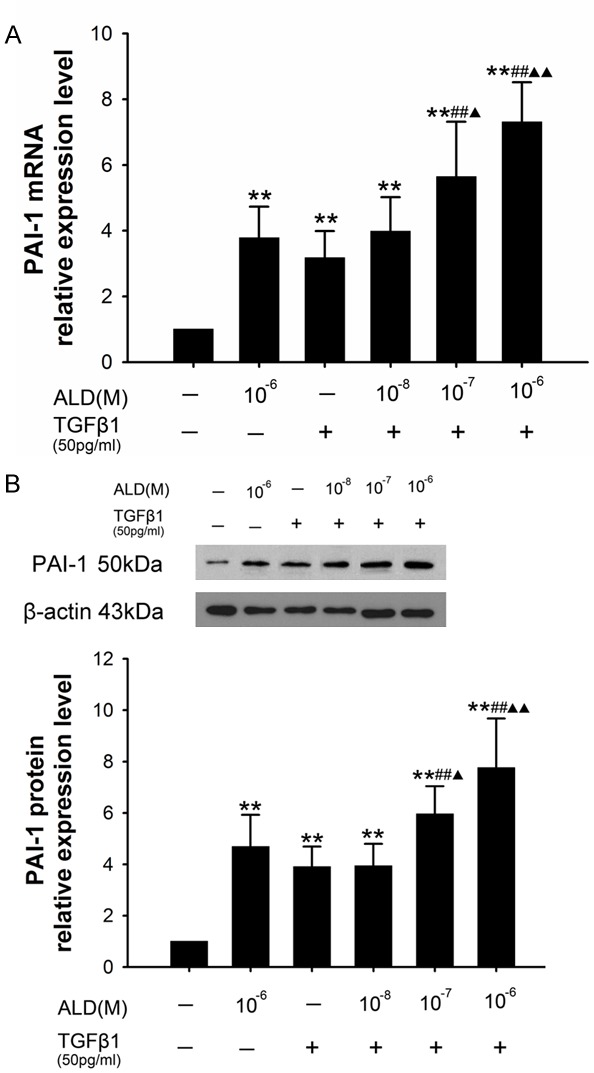
A. Effect of Ald and TGF-β1 on the PAI-1 mRNA expression in HSCs. B. Effect of Ald and TGF-β1 on the PAI-1 protein expression in HSCs. *vs control group, #10-7 M Ald+TGF-β1 group and 10-6 M Ald+TGF-β1 group vs TGF-β1 group, ▲10-7 M Ald+TGF-β1 group and 10-6 M Ald+TGF-β1 group vs ALD group. *P<0.05, **P<0.01, ***P<0.001.
Statistical analysis
Statistical analysis was performed with SPSS version 17.0. Quantitative data are expressed as mean ± standard deviation. Comparisons were done with t test between two groups and analysis of variance among groups. A value of P<0.05 was considered statistically significant.
Discussion
HSCs are major cells regulating ECM deposition in the liver. HSCs may regulate the uPA and PAI expressions to modulate the activities of plasmin and MMP family members, and then to maintain the balance between ECM synthesis and degradation. This is one of important ways in which HSCs regulate the ECM metabolism. Our results showed Ald treated HSCs had significantly increased PAI-1 synthesis and makredly elevated PAI-1 expression as compared to control group (P<0.01). These results demonstrate that Ald may directly induced PAI-1 over-expression but SPI may partially inhibit the Ald-induced PAI-1 expression. Ald acts on HSCs to modulate the fibrotic matrix protein metabolism. PAI-1 is a key factor modulating the fibrolysis and ECM deposition, and the Ald induced matrix degradation might be PAI-1 dependent. Our results indicated that Ald could also reduce the ECM degradation to induce hepatic fibrosis except for it physiologically functioning to maintain the dynamic balance of extracellular salt, water and potassium. In addition, this study also revealed that, besides increasing PAI-1 expression, Ald was able to elevate the secretion of type I cillagen in HSCs, and Ald and TGF-β1 could synergistically promote the production of type I collagen in HSCs as compared to treatment wth Ald alone, which however was blocked by SPI. There is evidence [18] showing that aldosterone/salt treated rats presented significantly increased expression of collagens in the kidney, accompanied by elevation of PAI-1 expression. Furthermore, the glomerular hypertrophy was significantly improved when there was PAI-1 deficiency. In addition, addition of MR blocker was able to reduce the PAI-1 expression and improve glomerulosclerosis in rats with diabetes or nephritis [20,21]. Our findings were consistent with results in previous studies: Ald is able to induce PAI-1 expression in HSCs, and SPR inhibits the bioeffects of Ald on HSCs.
Several studies have shown that Ald is able to increase TGF-β1 secretion in vitro, but little is known about TGF-β1 mRNA expression after Ald treatment [7,17,22]. In recent years, investigators focus on the effect of Ald on the TGF-β1 in HSCs in the absence of RAAS. Our previous study [23] showed Ald could induce the TGF-β1 expression in HSCs in vitro as compared to control group (P<0.05), and the TGF-β1 expression increased over time, but Ald in combination with SPI significantly reduced TGF-β1 expression as compared to treatment with Ald alone (P<0.05).
PAI-1 involves in some pathophysiological processes including fibrinolysis, recycling of ECM, fibrosis of tissues and organs and tissue repair after trauma. Thus, studies on the regulation of PAI-1 expression may provide new ways and methods for the therapy of related diseases. In recent years, there is evidence showing that PAI-1 plays multiple roles in the pathogenesis of chronic liver diseases. On one hand, PAI-1 induced inflamamtion and firbosis mediate the destruction of tissues, and on the other hand PAI-1 is able to help the liver repair after liver injury [24]. Our results showed Ald treated HSCs had elevated PAI-1 expression, and Ald (10-6 M) could exert synergistical effect with TGF-β1 to induce PAI-1 expression in HSCs (P<0.01 vs 10-6 M Ald group). Although the PAI-1 expression increased in HSCs after treatment with Ald (<10-6 M) and TGF-β1, there was no marked difference between Ald (<10-6 M) and TGF-β1 group and Ald (10-6 M) group. Thus, in our study, HSCs treated with Ald and TGF-β1 showed significantly increased PAI-1 expression as compared to Ald group (P<0.01). This suggests that Ald and TGF-β1 may synergistically induce PAI-1 expression in HSCs. There is evidence showing that Smad signaling pathway is crucial for the TGF-β1 mediated PAI-1 expression. Studies also reveal that TGF-β1 is effective to increase the transcriptional activity of PAI-1, and TGF-β1 may induce the formation of smad/Spl complex to activate Sp1 and up-regulate PAI-1 expression [25,26]. Our results also showed Ald significantly induced Smad in HSCs. Thus, we speculate that Ald induced PAI-1 expression is partially mediated by TGF-β1, which is consistent with findings from a study on pre-integrin β6 deficient mice [27].
Of note, available studies confirm that TGF-β1 over-expression is present in tissues with fibrosis [17,28]. Ald may interact with TGF-β to induce PAI-1 over-expression and then inhibit ECM degradation. Ald binds to MR to induce TGF-β1 expression and activation, leading to the increase in PAI-1 expression, and TGF-β1 neutralizing antibody may reduce the Ald induced PAI-1 expression, but it may not reduce to normal level. This indicates that Ald can stimulate PAI-1 expression in a TGF-β1 dependent manner, but this is not a unique pathway [29]. This synergistic effect may be observed in patients with heart failure or diabetic nephropathy whom are treated with ang II receptor blockers and/or aldosterone blockers [28,30-32] as well as animals with hypertension or glomerulonephritis [33-35]. In vitro experiments showed the TGF-β1 and FN expressions increased in Ald treated mesangial cells, which was antagonized by SPI. Our in vitro experiments also indicated that Ald induced a slight increase in PAI-1 expression in HSCs, and Ald and TGF-β1 could synergistically increase PAI-1 expression. These findings demonstrate that Ald may involve the fibrosis independent of RAAS, and can regulate TGF-β1 signaling pathway to up-regulate collagen expression and reduce MMP release, leading to the proliferation of fibroblasts.
Taken together, our findings indicate that Ald is able to induce PAI-1 over-expression in HSCs, which is partially mediated by TGF-β1. Increased PAI-1 expression may reduce the ECM degradation. Although Ald alone only induces slight increase in PAI-1 expression, Ald may synergistically exert effect with TGF-β1 to significantly induce PAI-1 expression, leading to the occurrence and development of fibrosis. SPI may partially inhibit the PAI-1 over-expression secondary to Ald treatment. Thus, PAI-1 may become a new target for the therapy of hepatic fibrosis. However, the specific signaling pathway involved in the Ald-induced hepatic fibrosis, the interaction among signal molecules and whether there is cross-talk among other signaling pathways are still poorly understood, and more studies are required to elucidate these issues.
Acknowledgements
Supported by Liver Fibrosis Research Foundation of Wang Bao En (No: CFHPC20131042) and the liver disease research fundation of Tianqing (No. TQGB20140148).
Disclosure of conflict of interest
None.
References
- 1.Connell JM, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186:1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 2.Takebayashi K, Matsumoto S, Aso Y, Inukai T. Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J Clin Endocrinol Metab. 2006;91:2214–7. doi: 10.1210/jc.2005-1718. [DOI] [PubMed] [Google Scholar]
- 3.Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–64. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- 4.Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, Chander PN, Ding G, Singhal PC. Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol. 2008;295:F73–81. doi: 10.1152/ajprenal.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patni H, Mathew JT, Luan L, Franki N, Chander PN, Singhal PC. Aldosterone promotes proximal tubular cell apoptosis: role of oxidative stress. Am J Physiol Renal Physiol. 2007;293:F1065–71. doi: 10.1152/ajprenal.00147.2007. [DOI] [PubMed] [Google Scholar]
- 6.Han JS, Choi BS, Yang CW, Kim YS. Aldosterone-induced TGF-beta1 expression is regulated by mitogen-activated protein kinases and activator protein-1 in mesangial cells. J Korean Med Sci. 2009;24(Suppl):S195–203. doi: 10.3346/jkms.2009.24.S1.S195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai L, Chen J, Hao CM, Lin S, Gu Y. Aldosterone promotes fibronectin production through a Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem Biophys Res Commun. 2006;348:70–5. doi: 10.1016/j.bbrc.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 8.Juknevicius I, Segal Y, Kren S, Lee R, Hostetter TH. Effect of aldosterone on renal transforming growth factor-beta. Am J Physiol Renal Physiol. 2004;286:F1059–62. doi: 10.1152/ajprenal.00202.2003. [DOI] [PubMed] [Google Scholar]
- 9.Zhou G, Kandala JC, Tyagi SC, Katwa LC, Weber KT. Effects of angiotensin II and aldosterone on collagen gene expression and protein turnover in cardiac fibroblasts. Mol Cell Biochem. 1996;154:171–8. doi: 10.1007/BF00226785. [DOI] [PubMed] [Google Scholar]
- 10.Fogo AB. Progression and potential regression of glomerulosclerosis. Kidney Int. 2001;59:804–19. doi: 10.1046/j.1523-1755.2001.059002804.x. [DOI] [PubMed] [Google Scholar]
- 11.Ma LJ, Nakamura S, Whitsitt JS, Marcantoni C, Davidson JM, Fogo AB. Regression of sclerosis in aging by an angiotensin inhibition-induced decrease in PAI-1. Kidney Int. 2000;58:2425–36. doi: 10.1046/j.1523-1755.2000.00426.x. [DOI] [PubMed] [Google Scholar]
- 12.Brown NJ, Nakamura S, Ma L, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, Vaughan DE, Fogo AB. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000;58:1219–27. doi: 10.1046/j.1523-1755.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- 13.Krag S, Osterby R, Chai Q, Nielsen CB, Hermans C, Wogensen L. TGF-beta1-induced glomerular disorder is associated with impaired concentrating ability mimicking primary glomerular disease with renal failure in man. Lab Invest. 2000;80:1855–68. doi: 10.1038/labinvest.3780196. [DOI] [PubMed] [Google Scholar]
- 14.Lund LR, Riccio A, Andreasen PA, Nielsen LS, Kristensen P, Laiho M, Saksela O, Blasi F, Danø K. Transforming growth factor-beta is a strong and fast acting positive regulator of the level of type-1 plasminogen activator inhibitor mRNA in WI-38 human lung fibroblasts. EMBO J. 1987;6:1281–6. doi: 10.1002/j.1460-2075.1987.tb02365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chun TY, Chander PN, Kim JW, Pratt JH, Stier CT Jr. Aldosterone, but not angiotensin II, increases profibrotic factors in kidney of adrenalectomized stroke-prone spontaneously hypertensive rats. Am J Physiol Endocrinol Metab. 2008;295:E305–12. doi: 10.1152/ajpendo.00512.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malgorzewicz S, Skrzypczak-Jankun E, Jankun J. Plasminogen activator inhibitor-1 in kidney pathology (Review) Int J Mol Med. 2013;31:503–10. doi: 10.3892/ijmm.2013.1234. [DOI] [PubMed] [Google Scholar]
- 17.Yuan J, Jia R, Bao Y. Aldosterone up-regulates production of plasminogen activator inhibitor-1 by renal mesangial cells. J Biochem Mol Biol. 2007;40:180–8. doi: 10.5483/bmbrep.2007.40.2.180. [DOI] [PubMed] [Google Scholar]
- 18.Ma J, Weisberg A, Griffin JP, Vaughan DE, Fogo AB, Brown NJ. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int. 2006;69:1064–72. doi: 10.1038/sj.ki.5000201. [DOI] [PubMed] [Google Scholar]
- 19.Usalan C, Buyukhatipoglu H. A dynamic comparative study concerning the effects of angiotensin-converting enzyme inhibitors and aldosterone receptor blockers on the fibrinolytic system. Clin Appl Thromb Hemost. 2008;14:203–9. doi: 10.1177/1076029607303614. [DOI] [PubMed] [Google Scholar]
- 20.Fujisawa G, Okada K, Muto S, Fujita N, Itabashi N, Kusano E, Ishibashi S. Spironolactone prevents early renal injury in streptozotocin-induced diabetic rats. Kidney Int. 2004;66:1493–502. doi: 10.1111/j.1523-1755.2004.00913.x. [DOI] [PubMed] [Google Scholar]
- 21.Gullulu M, Akdag I, Kahvecioglu S, Filiz G, Savci V. Aldosterone blockage in proliferative glomerulonephritis prevents not only fibrosis, but proliferation as well. Ren Fail. 2006;28:509–14. doi: 10.1080/08860220600779033. [DOI] [PubMed] [Google Scholar]
- 22.Han KH, Kang YS, Han SY, Jee YH, Lee MH, Han JY, Kim HK, Kim YS, Cha DR. Spironolactone ameliorates renal injury and connective tissue growth factor expression in type II diabetic rats. Kidney Int. 2006;70:111–20. doi: 10.1038/sj.ki.5000438. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Zhang Z, Zhu X, Wu H, Gao H, Yang C. Effect of aldosterone and its antagonist on the expression of PAI-1 and TGF-beta1 in rat hepatic stellate cells. Int J Clin Exp Med. 2014;7:4677–85. [PMC free article] [PubMed] [Google Scholar]
- 24.von Montfort C, Beier JI, Kaiser JP, Guo L, Joshi-Barve S, Pritchard MT, States JC, Arteel GE. PAI-1 plays a protective role in CCl4-induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol Gastrointest Liver Physiol. 2010;298:G657–66. doi: 10.1152/ajpgi.00107.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YQ, Sloan-Lancaster J, Berg DT, Richardson MA, Grinnell B, Tseng-Crank J. Differential mechanisms of plasminogen activator inhibitor-1 gene activation by transforming growth factor-beta and tumor necrosis factor-alpha in endothelial cells. Thromb Haemost. 2001;86:1563–72. [PubMed] [Google Scholar]
- 26.Datta PK, Blake MC, Moses HL. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-beta -induced physical and functional interactions between smads and Sp1. J Biol Chem. 2000;275:40014–9. doi: 10.1074/jbc.C000508200. [DOI] [PubMed] [Google Scholar]
- 27.Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, Sheppard D, Fogo AB. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(-/-) mice. Am J Pathol. 2003;163:1261–73. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu ZH, Yang ZC, Chen L, Lv ZD, Yi MJ, Ran N. Inhibition airway remodeling and transforming growth factor-beta1/Smad signaling pathway by astragalus extract in asthmatic mice. Int J Mol Med. 2012;29:564–8. doi: 10.3892/ijmm.2011.868. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Huang Y, Border WA, Noble NA. Perspectives on blockade of TGFbeta overexpression. Kidney Int. 2006;69:1713–4. doi: 10.1038/sj.ki.5000260. [DOI] [PubMed] [Google Scholar]
- 30.Chrysostomou A, Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N Engl J Med. 2001;345:925–6. doi: 10.1056/NEJM200109203451215. [DOI] [PubMed] [Google Scholar]
- 31.Rossignol P, Cleland JG, Bhandari S, Tala S, Gustafsson F, Fay R, Lamiral Z, Dobre D, Pitt B, Zannad F. Determinants and consequences of renal function variations with aldosterone blocker therapy in heart failure patients after myocardial infarction: insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study. Circulation. 2012;125:271–9. doi: 10.1161/CIRCULATIONAHA.111.028282. [DOI] [PubMed] [Google Scholar]
- 32.Rachmani R, Slavachevsky I, Amit M, Levi Z, Kedar Y, Berla M, Ravid M. The effect of spironolactone, cilazapril and their combination on albuminuria in patients with hypertension and diabetic nephropathy is independent of blood pressure reduction: a randomized controlled study. Diabet Med. 2004;21:471–5. doi: 10.1111/j.1464-5491.2004.01194.x. [DOI] [PubMed] [Google Scholar]
- 33.Asai M, Monkawa T, Marumo T, Fukuda S, Tsuji M, Yoshino J, Kawachi H, Shimizu F, Hayashi M, Saruta T. Spironolactone in combination with cilazapril ameliorates proteinuria and renal interstitial fibrosis in rats with anti-Thy-1 irreversible nephritis. Hypertens Res. 2004;27:971–8. doi: 10.1291/hypres.27.971. [DOI] [PubMed] [Google Scholar]
- 34.Kramer AB, van der Meulen EF, Hamming I, van Goor H, Navis G. Effect of combining ACE inhibition with aldosterone blockade on proteinuria and renal damage in experimental nephrosis. Kidney Int. 2007;71:417–24. doi: 10.1038/sj.ki.5002075. [DOI] [PubMed] [Google Scholar]
- 35.Onozato ML, Tojo A, Kobayashi N, Goto A, Matsuoka H, Fujita T. Dual blockade of aldosterone and angiotensin II additively suppresses TGF-beta and NADPH oxidase in the hypertensive kidney. Nephrol Dial Transplant. 2007;22:1314–22. doi: 10.1093/ndt/gfl780. [DOI] [PubMed] [Google Scholar]