

Abstract
This work aimed at determining the ideal ischemia time in an in vitro ischemia-reperfusion model of spinal cord injury. Rat spinal cord slices were prepared and then exposed or not to oxygen deprivation and low glucose (ODLG) for 30, 45, 60, 75 and 90 minutes. Cell viability was assessed by triphenyltetrazolium (TTC), lactate dehydrogenase (LDH) release, and fluorochrome dyes specific for cell dead (ethidium homodimer) using the apotome system. Glutamate release was enzymatically measured by a fluorescent method. Gene expression of apoptotic factors was assessed by real time RT-PCR. Whereas spinal cord slices exposed to ODLG exhibited mild increase in fluorescence for 30 minutes after the insult, the 45, 60, 75 and 90 minutes caused a 2-fold increase. ODLG exposure for 45, 60, 75 or 90 minutes, glutamate and LDH release were significantly elevated. nNOS mRNA expression was overexpressed for 45 minutes and moderately increased for 60 minutes in ODLG groups. Bax/bcl-xl ratio, caspase 9 and caspase 3 mRNA expressions were significantly increased for 45 minutes of ODLG, but not for 30, 60, 75 and 90 minutes. Results showed that cell viability reduction in the spinal cord was dependent on ischemic time, resulting in glutamate and LDH release. ODLG for 45 minutes was adequate for gene expression evaluation of proteins and proteases involved in apoptosis pathways.
Keywords: Apoptosis, oxygen deprivation low glucose, spinal cord ischemia, cell viability, excitotoxicity
Introduction
Ischemic spinal cord injuries are observed in acute mechanical traumas and secondary to vascular and tissue insult, leading to irreversible lesions [1,2]. Clinical data indicate that in addition to typical traumatic causes of acute spinal cord lesions, such as automobilistic or firearm accident, there is a growing etiopathology importance of non-traumatic injuries (39%), including those caused by spinal ischemia, in particular paraplegia which affects up to 22% of patients undergoing thoraco-abdominal aneurysm surgery [3].
Spinal cord ischemia results in depletion of highly energetic compounds by metabolic mechanisms dependents on energy in neurons and glial cells, since the altered glucose transport turns inadequate the available substract. Consequently, ischemic insults interrupt deoxyribonucleic acid (DNA) synthesis, as well as the synthesis and transport of lipids and protein, neuronal death and damage to synaptogenesis and mielinization. Such phenomena are not exclusive of the acute phase of hypoxic event, but they also encompass the reperfusion process [4-7].
Ischemia-reperfusion experimental models have been developed in many animal species (rats, cats, rabbits) by using some in vivo procedures, such as balloon compression and partial or total arteries occlusion [8-10]. Nonetheless, the complexities of in vivo system associated with the multiplicity of the disease process may impair the isolated interpretation of the pathophysiological and pharmacological mechanisms. Thus, organotypic slice model can overlap the limitations of in vivo models, providing more control of experimental situation for the assessment of therapeutic approaches [11-13]. However, what would be the ideal ischemia time for a model of in vitro spinal cord ischemia? Some studies showed that 10 min of ischemia in hippocampus [13,14] and 45 min in retina [15] were sufficient to cause cell death and excessive glutamate release, but until this moment, there is no information on the literature regarding ischemia time in spinal cord slices.
Organotypic spinal cord slice can be utilized for assessing the various cell injury conditions and ischemic insults. The majority of investigations used organotypical spinal cord cultures prepared from embryonic or newborn rats and mice in their studies. Although such model promotes long lasting embryonic and newborn cultures [16-18], it is highly expensive. Besides, there is a significant difference in function, circuitry connections, and regenerative capacity in the immature spinal cord compared to adults’, which turns out to be a disadvantage.
This work aimed to determine the ideal ischemia time that activates primary and secondary mechanisms of cell death in an in vitro ischemia-reperfusion model of spinal cord injury. For so, this study compared different ischemia times on rat spinal cord slices exposed to oxygen deprivation and low glucose (ODLG), followed by a reoxygenation period. Insult intensity was evaluated through cell viability, excitotoxicity and gene expression of apoptotic factors.
Materials and methods
Spinal cord slices and procedure perfusion chamber
All procedures were approved by the local Ethics Committee (CETEA/UFMG no 14/2010) and followed the guidelines for the Use and Care of Animals for Research. Wistar rats (180-200 g) were killed by decapitation, their spinal cord rapidly (< 1 min) removed by hydraulic extrusion and submerged in cold (4°C) artificial cerebrospinal fluid (ACSF) containing (in mMol/L): 127 NaCl, 2 KCl, 10 glucose, 1.2 KH2PO4, 26 NaHCO3, 2 MgSO4, 2 CaCl2, 13 HEPES bubbled with 95% O2/5% CO2 and 7.4 pH [19,20]. Lumbossacral regions were dissected and transverse slices (0.4 mm) were obtained with a McIlwain Tissue Chopper (Brinkman Instruments, UK). The slices were placed on nylon mesh platforms of 12 covered incubation chambers (3 slices/chamber) of a Brandel Suprafusion System SF-12 (Gaithersburg, MD, USA). In every procedure, the rate perfusion was 0.5 mL/min. The slices were submerged on ACSF, bubbled with 95% O2/5% CO2 for 90 min at 37°C to recover from the mechanical trauma. After this period, slices submitted to the ODLG insult were perfused with ACSF with 4 mmol/L glucose for 30, 45, 60, 75 and 90 minutes bubbled with 95% N2/5% CO2, except control conditions. Thereafter, the spinal cord slices were washed with ACSF with 10 mM glucose and 95% O2/5% CO2 during 4 hours (reperfusion period) before assessing cell viability.
Assessment of cell death
To analyze cell death, slices were stained with 2 mmol/L ethidium homodimer-1 (live/dead assay, Molecular Probes, Eugene, OR) for 30 min and then washed for 15 min with 95% O2/5% CO2 ACSF with 10 mM glucose at room temperature. During the staining procedure, slices were protected from light.
Images were acquired in a Zeiss Axiovert 200 M Microscope, using the Apotome system in order to obtain optical sections from a Z-series at 0.02 mm interval. Objectives used were 20x dry. Image J software was used to combine consecutive optical sections from a Z-series and create image constructions. All nuclei in this whole field were counted. Quantification of dead cells required identification of their nuclei fluorescently stained with ethidium homodimer, which was related to ischemia-induced neurotoxicity. Morphological analysis of dead cells was performed in eight fields of the ventral horn (two fields/slice) from five different experiments in ischemic and non-ischemic conditions.
Triphenyltetrazolium (TTC) assay
Cell viability was determined by converting 2,3,5-TTC to the insoluble formazan. The reduction of this compound is dependent on mitochondrial respiratory activity. Therefore, this conversion can be proportional to the number of viable cells. After decapitation of the animals, the spinal cord section was subjected to 400 mM with the aid of a tissue cutter and after processing in the perfusion chamber, the slices were incubated at 37°C in a solution of 2% TTC for 90 minutes. After this period, the TTC was removed and the tissue washed with 0.9% saline solution and then added 1.5 mL of a solution containing dimethylsulfoxide (DMSO) and ethanol in a 1:1 ratio and incubated for 24 hours at room temperature, protected from light, to solubilize the formazan. The reading was held in a spectrophotometer at 485 nm and results normalized by the weight of tissue, in torsion balance.
LDH release assay
After ODLG insult, the incubation medium containing spinal cord slices and the supernatant were collected for LDH assay (LDH Liquiform, Labtest S.A., MG, Brazil). Protein in the pellet was measured as described by Bradford [21]. LDH measurement in the supernatant was performed enzymatically catalyzed in the interconversion of pyruvate and lactate, in the presence of NADH. The reduction of the absorbance at 340 nm, as consequence of NADH oxidations, was proportional to the LDH activity in the sample. Each experiment consisted of at least two replicates per condition. LDH level was calculated as U/L/mg of protein.
Glutamate release assay
After ODLG insult, the incubation medium containing spinal cord slices and the supernatant were collected for glutamate assay. Protein in the pellet was measured as described by Bradford [21]. Glutamate measurement in the supernatant was performed enzymatically after the increase in the fluorescence due to the NADPH+ production in the presence of glutamate dehydrogenase and NADP+ [22]. To start the assay, NADP+ (1.0 mM) and glutamate dehydrogenase (50 units) were added to the samples and the emitted fluorescence was measured [22,23]. The excitation wavelength was set at 360 nm and the emission wavelength to 450 nm using a Shimadzu RF-5301PC spectrofluorimeter (Kyoto, Japan). Glutamate level was calculated as pmol of glutamate per milligram of protein. Each experiment consisted of at least two replicates per condition.
RNA extraction and real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA), chloroform, and isopropanol. The precipitate was washed in ethanol, air-dried, and re-diluted in diethyplyrocarbonate (DEPC)-treated distilled water. The amount and purity of extracted RNA was quantified by spectrophotometry (GeneQuantTM pro RNA/DNA; GE Healthcare, Piscataway, NJ, USA). RNA reverse transcription and real time-PCR reactions were performed with SuperScriptTM III Platinum® Two-Step qRT-PCR Kit with SYBR Green (Invitrögen, Carlsbad, CA, USA). Primers sequences are shown in Table 1. For real-time PCR, data were analyzed with 7500 software v.2.0.1 Applied Biosystems, using the comparative Cycle threshold (Ct) method [24]. mRNA level is presented as number of copies per 103 copies of β-actin mRNA; considering n = 3.3 Ct, and 10n = difference in the number of mRNA copies.
Table 1.
Sequences of primers for the real time RT-PCR analysis
| Gene | Access number | Primer sequence (5’-3’) | Product size (bp) |
|---|---|---|---|
| Bcl-xl | NM_001033670.1 | Forward: 5’CCCCAGAAGAAACTGAACCA 3’ | 300 |
| Reverse: 5’AGTTTACCCCATCCCGAAAG 3’ | |||
| Bax | NM_017059.1 | Forward: 5’CCAAGAAGCTGAGCGAGTGTCTC 3’ | 147 |
| Reverse: 5’AGTTGCCATCAGCAAACATGTCA 3’ | |||
| Caspase-9 | NM_031632.1 | Forward: 5’TGGAGGAGGCTGACCGGCAA 3’ | 77 |
| Reverse: 5’CCACAGCTCCGCGACTTGCA 3’ | |||
| Caspase-3 | NM_012922.2 | Forward: 5’TGGAGGAGGCTGACCGGCAA 3’ | 70 |
| Reverse: 5’CTCTGTACCTCGGCAGGCCTGAAT 3’ | |||
| Beta-actin | NM_031144.2 | Forward: 5’GCGTCCACCCGCGAGTACAA 3’ | 118 |
| Reverse: 5’ACATGCCGGAGCCGTTGTCG 3’ | |||
| nNOS | NM_052799 | Forward: 5’TCCCTCTAGCCAAAGAATTTCTCG 3’ | 120 |
| Reverse: 5’GGTAGGTGCTGGTGCTTTCAA 3’ |
Statistical analysis
All values are expressed as means ± SEM. Data were analyzed through one-way ANOVA and Student-Newman Keuls post hoc test. P < 0.05 values were considered statistically significant.
Results
Cell viability
For TTC technique, there was no significant decrease in cell viability slices of spinal cord subjected to 30-minute ischemic insult. For 45 minutes, there was a decrease in mitochondrial activity compared to the control group and ODLG 60 there was no further increase in tissue damage. Cell death was 17.43, 58.40, 80.07, 80.17 and 85.39% in ODLG 30, 45, 60, 75, and 90 minutes, respectively, compared to the control group (Figure 1).
Figure 1.
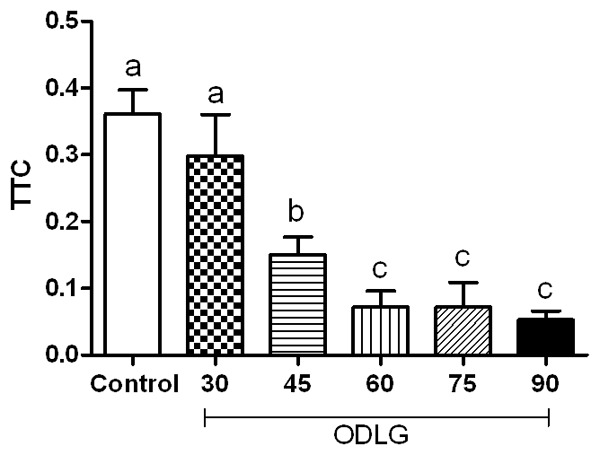
Cell viability by TTC assay. Mean ± SEM in all experimental groups. Means with different letters differed significantly (P < 0.05).
Compared to non-ischemia group, spinal cord slices exposed to ODLG exhibited significant increase in fluorescence at 30 minutes after the insult (P < 0.05), but mild increase in tissue injury, whereas 45 minutes exposure caused a 2-fold increase in fluorescence (P < 0.05). The mean number of nuclei/field ± SEM was: 55.96±3.5; 72.70±2.6; 103±6.1; 124.7±8.1; 132.2±12.5 e 172.6±18.4 for control, ODLG 30, ODLG 45, ODLG 60, ODLG 75, and ODLG 90 min groups, respectively. These results showed cell injury of 23.0; 45.7; 40.6; 57.7 and 67.6%, on ODLG 30, 45, 60, 75 and 90 min, respectively, compared to control group. Although cell injury was significant for all ischemia times, it reached a saturated level in response to ODLG for groups 45, 60, 75 and 90 min (Figure 2). The spinal cord slice on control group did not differ from slice analyzed immediately after chopping the spinal cord (sham group). This result suggests that cell death in this condition is related to mechanical trauma and not to the perfusion in the chamber (P > 0.05).
Figure 2.
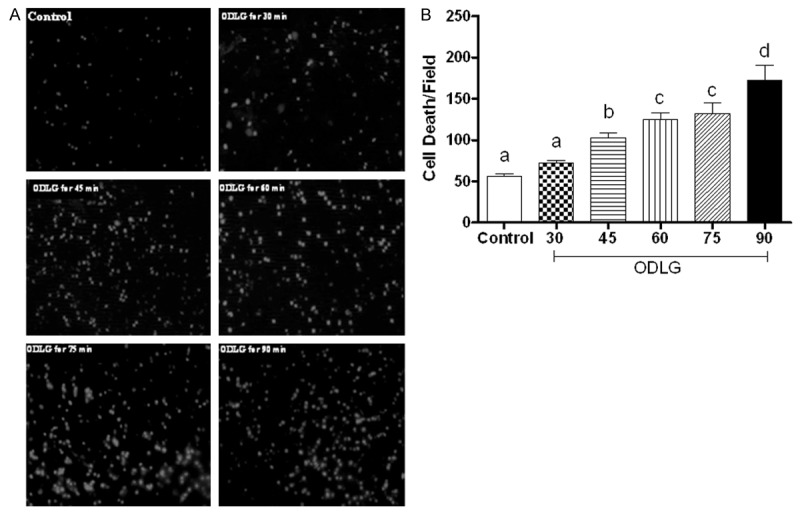
Induction of cell death by different times of ischemia-reperfusion. Spinal cord slices were stained with ethidium homodimer-1 after been submitted to slicing (sham), perfusion without ischemia (control), and oxygen deprivation and low glucose (ODLG) of 30, 45, 60, 75 and 90 minutes. A. Photomicrographs of ventral horns of the lumbossacral spinal cord showing fluorescent dead cells in all experimental groups. Scale bar, 50 μm. B. Mean ± SEM number of dead cells per field in all experimental groups. Means with different letters differed significantly (P < 0.05).
Glutamate and LDH release during ischemic insult
The mean glutamate concentration ± SEM was: 23.65±3.6; 34.38±2.8; 45.69±2.6; 48.97±2.1; 61.83±5.0; 59.10±9.3 for control, ODLG 30, ODLG 45, ODLG 60, ODLG 75, and ODLG 90 min groups, respectively (Figure 3). After ischemic insult of 45, 60, 75 or 90 min, glutamate concentration was markedly and significantly elevated. There was no statistical difference between the effect of 45 and 60 min on the release of glutamate induced by ischemia (Figure 3, P > 0.05). Therefore, LDH release for 30 minutes of ODLG was similar to that of control conditions, but it was statistically significant for 45, 60, 75 and 90 minutes of ODLG (Figure 4, P < 0.05).
Figure 3.
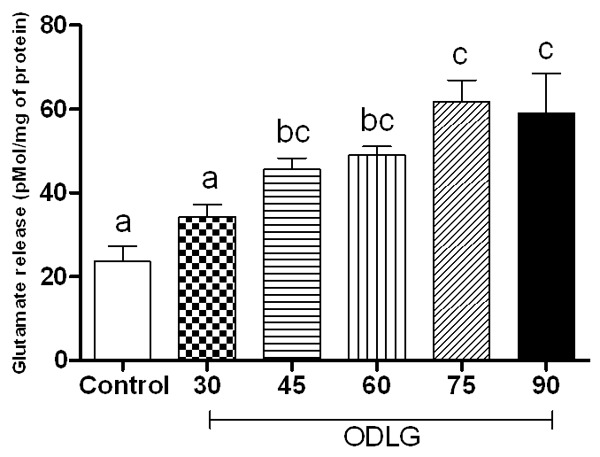
Glutamate release induced by perfusion without ischemia (control), and oxygen deprivation and low glucose (ODLG) at 30, 45, 60, 75 and 90 minutes. Data shown are mean ± SEM from five different experiments. Means with different letters differed significantly (P < 0.05).
Figure 4.
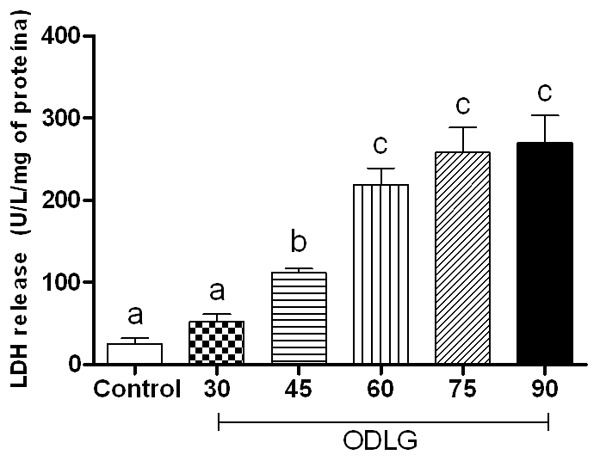
LDH release induced by perfusion without ischemia (control), and oxygen deprivation and low glucose (ODLG) at 30, 45, 60, 75 and 90 minutes. Data shown are mean ± SEM from five different experiments. Means with different letters differed significantly (P < 0.05).
Caspase 9, caspase 3, nNOS, Bax and Bcl-xl mRNA levels after ischemic reperfusion insult in rat spinal cord slices
The nNOS mRNA expression was 0.39±0.13; 2.73±0.33; 8.17±1.34; 4.85±0.37; 1.41±0.20, and 0.99±0.87 in control, 30, 45, 60, 75 and 90 min ODLG, respectively. nNOS mRNA was overexpressed in 45 min and moderately increased in ODLG 60 (Figure 5).
Figure 5.
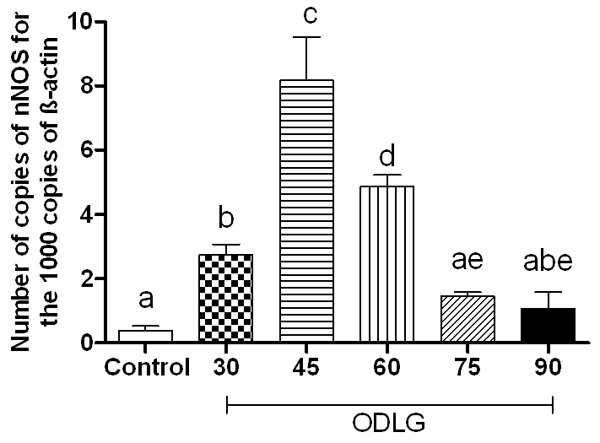
Quantitative nNOS mRNA expression through real time PCR in spinal cord slices submitted to perfusion without ischemia (control) and oxygen deprivation and low glucose (ODLG) of 30, 45, 60, 75 and 90 minutes. Data shown are mean mean ± SEM from three independent experiments. Means with different letters differed significantly (P < 0.05).
The Bax/Bcl-xl ratio mRNA expression was significantly increased at 45 and 60 min of ODLG, but not at 30, 75 and 90 min (Figure 5, P > 0.05). The mRNA expression values were: 4.48±1.46; 51.26 ±17.15; 37.33±5.316; 14.29±2.77 and 21.78±3.06 for control, 30, 45, 60, 75 and 90 min of ODLG, respectively (Figure 6). Comparable results were obtained using different set of rats.
Figure 6.
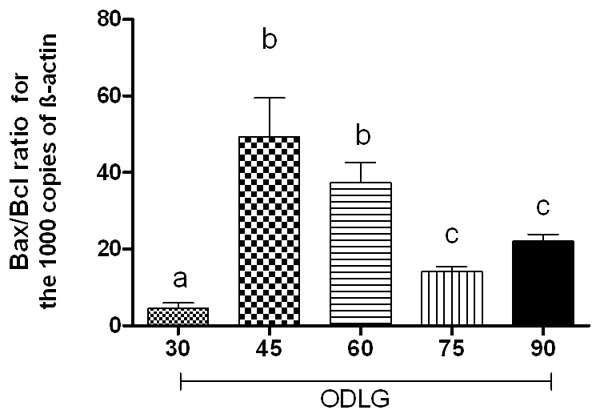
Quantitative Bax/Bcl-xl ratio mRNA expression through real time PCR in spinal cord slices submitted to oxygen deprivation and low glucose (ODLG) of 30, 45, 60, 75 and 90 minutes. Data shown are mean ± SEM from three independent experiments. Means with different letters differed significantly (P < 0.05).
Caspase 9 and caspase 3 mRNA expression (Figure 7) increased with 45 min of ODLG and 4 h of reperfusion (P < 0.05) compared to control conditions. For 60 min of ODLG, the expression of these proteases was similar to control and 30 min ODLG. Caspase 9 mRNA expression was 2.97±1.3; 7.15±1.6; 17.63±2.7; 4.58±2.0; 5.42±1.6, and 3.032±0.9 in control, and ODLG 30, 45, 60, 75, and 90 min, respectively. For caspase 3, mRNA expression was: 0.39±0.13; 2.73±0.33; 8.17±1.34; 4.85±0.37; 1.41±0.20 and 0.99±0.87 in control and ODLG 30, 45, 60, 75 and 90 min, respectively.
Figure 7.
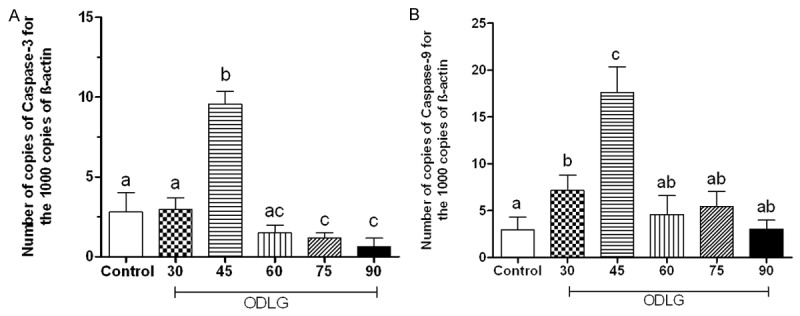
Quantitative caspase-9 (A) and caspase-3 (B) mRNA expression through real time PCR in spinal cord slices submitted to perfusion without ischemia (control) and oxygen deprivation and low glucose (ODLG) of 30, 45, 60, 75 and 90 minutes. Data shown are mean ± SEM. Means with different letters differed significantly (P < 0.05).
Discussion
As viability of isolated spinal cord is highly dependent on oxygenation, it is imperative to avoid or minimize tissue hypoxia during dissection. In order to achieve this, we used hydraulic extrusion technique for spinal cord dissection, which is a faster procedure (< 30 seconds), compared to conventional laminectomy. This procedure was used by other authors [20], and preservation of glial cells and neurons in white and grey matters were confirmed morphologically and electrophysiologically [20]. Additionally, it was used ACSF bubbled at 95% O2/5% CO2 and glucose during procedure, so that possible electrolyte imbalances might be reduced, avoiding excitotoxicity. The lumbossacral segment and the ventral horn were chosen based on recent studies that demonstrate a higher vulnerability of these regions towards ischemia/reperfusion [25-27,11].
Citotoxicity and cell death have been investigated through fluorescent markers such as ethidium homodimer, propidium iodide, sytox-green, 3-(4,5-dimethythiazol-2-yl)-2-5-diphenyltetrazolium bromide-MTT [12,28]. Ethidium homodimer has a high affinity for nucleic acids and is impermeable to cells with intact membranes. The membranes of dying cells become leaky and allow the etidium homodimer entry into the nucleus. Upon binding to DNA or RNA, the fluorescence intensity increases up to 40 fold in dying cells. This technique allows assessment of neuron viability in 0.04 mm thick slices without requiring fixation or ultrathin slicing [13].
Thirty minutes of ODLG caused meaning loss of cell viability, with no remarkable increase in LDH and glutamate release. However, 45, 60, 75 and 90 minutes caused progressive loss of cell viability compared to control and were accompanied by glutamate and LDH increase, and TTC decrease. Thus, it is possible to suggest that loss of cell viability is dependent on ischemic insult time, and is associated with glutamate and LDH release. However, the significant increase in LDH release in 60, 75 and 90 min ODLG group compared to 45 min indicates that necrotic death may have prevailed in this group. Furthermore, our study suggests that after the first four hours of spinal cord ischemia, glutamate-mediated excitotoxicity is the major contributor to the resulting glial and neuronal damage. Studies report that ischemic injuries associated with hypoxia, vascular disruption, and trauma can alter synaptic function, through increased storage of extracellular glutamate and excessive stimulation of glutamate receptors. Over stimulation of glutamate receptor/channel complex triggers calcium influx, leading to cell injury or death [1,29,30]. It means that, when intracellular calcium concentrations ([Ca2+]i) reach non-physiological levels, several mechanisms are activated to remove it. However, such systems fail or are insufficient during excitotoxicity secondary to ischemia. Unchecked influx of Ca2+ can trigger apoptotic as well as necrotic death [2,31,32].
The relation between excitotoxicity and cell death, as observed in the present study, was already described by others [1,29]. These authors cited different routes of calcium ion entries in the cell, with different responses during ischemia and showed that voltage-gated Ca2+ channels do not elicit cell death, whereas N-methyl-D-aspartate receptor (NMDAR), 2-amino-3 (3-hydroxy-5-methylisox-azol-4yl) propionate receptor (AMPAR), and kainate receptors, which are ionotropic, are associated with significant Ca2+ dependent toxicity.
The increase of cytosolic calcium activates calcium-dependent enzymes, including NOS, resulting in nitric oxide production. In the present study, the overexpression of nNOS in all groups submitted to ischemic insult (Figure 4) restates nitric oxide participation in the pathophysiology of ischemia as reported by other authors [26,32,33], and the significant increase in the expression of this gene in 45 min of ODLG group suggested that this ischemia time promotes increase of glutamate activates NMDA receptors and subsequently influences the glutamate-induced neuronal death. It is worth noting that one of nNOS regulators is the NMDA receptor, NR1 and NR2 subtypes [31]. When these receptors are stimulated, they activate nNOS, which may contribute to secondary damage after ischemic insults [34,35].
The evaluation of Bax/Bcl-xl ratio mRNA expression revealed activation of the intrinsic apoptosis pathway for the 45 min ODLG group, which was not observed in 30, 60, 75, and 90 min ODLG groups. The under expression of Bax/Bcl-xl indicates that 30 minutes ischemia was not sufficient to cause acute activation of the apoptosis pathway, and it is in accordance with the results of cell viability, glutamate, LDH, caspase 9, and caspase 3 for this group. For the 60, 75 and 90 min ODLG groups, the results of Bax/Bcl-xl ratio, caspase 9, and caspase 3 expressions suggest that apoptosis pathway was not activated in this ischemia time, and that cell death, as observed by ethidium homodimer stain, occurred through a necrosis process. The increase of Bax/Bcl-xl ratio after 45 min of ischemia promoted the release of cytocrome C and apoptosis-inducing factor (AIF) [36], which activated caspase 9 and subsequently, caspase 3, confirming that apoptosis was the main responsible by cell death in this group.
It is known that intensity and time of insult, and excitotoxicity, caused by glutamate release, may induce cell death by necrosis and/or apoptosis [1,13]. Studies report that in mild to moderate insult, cell death in rats’ cortical cell cultures took place by apoptosis, while in prolonged periods of insults, secondary necrosis prevailed-the true lesion nature. Such event can be related to loss of cell homeostasis by ionic imbalance with overload of intracellular calcium and energy loss, resulting in membrane lysis and neuronal DNA damage [37]. The observed results seems to reinforce that 60 min ischemia have compromised metabolic activity in spinal cord slices, probably through decreased ATP values and elevated aerobic glycosis [37].
In this proposed ischemic/reperfusion model, significant excitotoxicity was obtained after 45, 60, 75 and 90 minutes of ischemia, but early activation of the apoptotic pathway was only confirmed after 45 minutes of ODLG. Thus, this model can be a valuable tool to evaluate the neuroprotective effects of pharmacological compounds in a quick accurate and consistent manner.
Conclusion
Based on the evidences shown, it is possible to conclude that Cell viability reduction in the spinal cord was dependent on ischemia time, resulting in glutamate and LDH release. 45 minutes of ischemia and four hours of reperfusion were adequate for gene expression evaluation of proteins and proteases involved in apoptosis pathway. The proposed in vitro experimental model is viable and reproducible for studies of pathophysiologic mechanisms of ischemia/reperfusion.
Acknowledgements
CNPq and FAPEMIG grants supported this study. The authors also would like to inform that there is no any kind of conflict of interest that would prevent acceptance or publication of this paper.
Disclosure of conflict of interest
None.
References
- 1.Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451–464. doi: 10.1016/j.spinee.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Aslan A, Cemek M, Buyukokuroglu ME, Altunbas K, Bas O, Yurumez Y, Cosar M. Dantrolene can reduce secondary damage after spinal cord injury. Eur Spine J. 2009;18:1442–1451. doi: 10.1007/s00586-009-1033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Berg ME, Castellote JM, Mahillo-Fernandez I, de Pedro-Cuesta J. Incidence of spinal cord injury worldwide: a systematic review. Neuroepidemiology. 2010;210:184–192. doi: 10.1159/000279335. [DOI] [PubMed] [Google Scholar]
- 4.Gaviria M, Privat A, D’Arbigny P, Kamenka J, Haton H, Ohanna F. Neuroprotective effects of a novel NMDA antagonist, gacyclidine, after experimental contusive spinal cord injury in adult rats. Brain Res. 2000;874:200–209. doi: 10.1016/s0006-8993(00)02581-6. [DOI] [PubMed] [Google Scholar]
- 5.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. New Eng J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 6.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. 2007;4:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Price DL. New order for neurologic disorders. Nature. 1999;399:A3–A5. doi: 10.1038/399a003. [DOI] [PubMed] [Google Scholar]
- 8.Cayli SR, Ates O, Karadag N, Altinoz E, Yucel N, Yologlu S, Kocak A, Cakir CO. Neuroprotective effect of etomidate on functional recovery in experimental spinal cord injury. Int J Dev Neurosci. 2006;24:233–239. doi: 10.1016/j.ijdevneu.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Kolenda H, Steffens H, Hagena J, Schomburg ED. Different susceptibility of facilitatory and inhibitory spinal pathways to ischemia in the cat. Neurosci Res. 2003;47:357–66. doi: 10.1016/j.neures.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Yu Q, Zhou Q, Huang H, Wang Y, Tian S, Duan D. Protective effect of etomidate on spinal cord ischemia reperfusion injury induced by aortic occlusion in rabbits. Ann Vasc Surg. 2010;24:225–232. doi: 10.1016/j.avsg.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 11.Kolesárová M, Pavel J, Lukacova N, Kolesár D, Marsala J. Effects of ischemia in vivo and oxygen-glucose deprivation in vitro on NOS pools in the spinal cord: comparative study. Cel Mol Neurobiol. 2006;26:1281–1294. doi: 10.1007/s10571-006-9032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krassioukov A, Ackery A, Schartz G, Kolesár D, Marsala J. An in vitro model of neurotrauma in organotypic spinal cord cultures from adult mice. Brain Res Protoc. 2002;10:60–68. doi: 10.1016/s1385-299x(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 13.Monette R, Small D, Mealing G, Morley P. A fluorescence confocal assay to assess neuronal viability in brain slices. Brain Res Brain Res Protoc. 1998;2:99–108. doi: 10.1016/s1385-299x(97)00020-2. [DOI] [PubMed] [Google Scholar]
- 14.Pinheiro ACN, Silva AJ, Prado MA, Cordeiro MN, Richardson M, Batista MC, de Castro Junior CJ, Massensini AR, Guatimosim C, Romano-Silva MA, Kushmerick C, Gomez MV. Phoneutria spider toxins block ischemia-induced glutamate release, neuronal death, and loss of neurotransmission in hippocampus. Hippocampus. 2009;19:1123–1129. doi: 10.1002/hipo.20580. [DOI] [PubMed] [Google Scholar]
- 15.Agostini RM, Pinheiro ACN, Binda NS, Romanosilva MA, Cordeiro MN, Richardson M, Guimarães ALS, Gomez MV. Phoneutria spider toxins block ischemia-induced glutamate release and neuronal death of cell layers of the retina. Retina. 2011;3:1392–1399. doi: 10.1097/IAE.0b013e318205b249. [DOI] [PubMed] [Google Scholar]
- 16.Fujimoto S, Katsuki H, Kume T, Kaneko S, Akaike A. Mechanisms of oxygen glucose deprivation-induced glutamate release from cerebrocortical slice cultures. Neurosci Res. 2004;50:79–87. doi: 10.1016/j.neures.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Kerkut GA, Bagust J. The isolated mammalian spinal cord. Prog Neurobiol. 1995;46:1–48. doi: 10.1016/0301-0082(94)00055-m. [DOI] [PubMed] [Google Scholar]
- 18.Van Westerlaak MGH, Joosten EAJ, Gribnau AAM, Gribnau AA, Cools AR, Bär PR. Differential cortico-motoneuron vulnerability after chronic mitochondrial inhibition in vitro and the role of glutamate receptors. Brain Res. 2001;922:243–249. doi: 10.1016/s0006-8993(01)03178-x. [DOI] [PubMed] [Google Scholar]
- 19.Chery N, Yu XH, Koninck H. Visualization of lamina I of the dorsal horn in live adult rat spinal cord slices. J Neurosci Methods. 2000;96:133–142. doi: 10.1016/s0165-0270(99)00195-8. [DOI] [PubMed] [Google Scholar]
- 20.Moghaddasi M, Velumian AA, Zhang L, Fehlings MG. An ex vivo preparation of mature mice spinal Cord to study synaptic transmission on motoneurons. J Neurosci Methods. 2007;159:1–7. doi: 10.1016/j.jneumeth.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Nicholls DG, Sihra TS, Sanchez-Prieto J. Calcium dependent and independent release of glutamate from synatosomes monitored by continuous fluorometry. J Neurochem. 1987;49:50–57. doi: 10.1111/j.1471-4159.1987.tb03393.x. [DOI] [PubMed] [Google Scholar]
- 23.Romano-Silva MA, Ribeiro AM, Ribeiro-Santos R, Ribeiro AM, Gomez MV, Diniz CR, Cordeiro MN, Brammer MJ. Rat cortical synaptosomes have more than one mechanism for calcium entry linked to rapid glutamate release: Studies using the Phoneutria nigriventer toxin PhTx2 and potassium depolarization. Biochem J. 1993;269:313–319. doi: 10.1042/bj2960313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi T, Sakurai M, Abe K, Sadahiro M, Tabayashi K, Itoyama Y. Apoptosis of motor neurons with induction of caspase in the spinal cord after ischemia. Stroke. 2010;29:1007–1013. doi: 10.1161/01.str.29.5.1007. [DOI] [PubMed] [Google Scholar]
- 26.Kolesár D, Kolesarova M, Pavel J, Dávidová A, Marsala J, Lukácová N. Region-specific sensitivity of the spinal cord to ischemia/reperfusion: the dynamic of changes in catalytic NOS activity. J Physiol Sci. 2009;59:97–103. doi: 10.1007/s12576-008-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nohda K, Nakatsuka MD, Takeda D, Miyazaki N, Nishi H, Sonobe H, Yoshida M. Selective vulnerability to ischemia in the rat spinal cord. Spine. 2007;10:1060–1066. doi: 10.1097/01.brs.0000261560.53428.90. [DOI] [PubMed] [Google Scholar]
- 28.Noraberg J, Kristensen BW, Zimmet J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc. 1999;3:278–290. doi: 10.1016/s1385-299x(98)00050-6. [DOI] [PubMed] [Google Scholar]
- 29.Hagg T, Oudega M. Degenerative and spontaneous regenerative processes after spinal cord injury. J Neurotrauma. 2007;23:264–280. doi: 10.1089/neu.2006.23.263. [DOI] [PubMed] [Google Scholar]
- 30.Rose CR, Rose SG, Waxman R, Ranson R. Effects of glucose deprivation, chemical hypoxia, simulated ischemia on Na+ homeostasis in rat spinal cord astrocytes. J Neurosci. 1998;18:3554–3562. doi: 10.1523/JNEUROSCI.18-10-03554.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Zhao YN, Yang EB, Ling EA, Wang Y, Hassouna MM, Mack P. Induction of neuronal and inducible nitric oxide synthase in the motoneurons of spinal cord following transient aorta occlusion in rats. J Surg Res. 1999;87:185–193. doi: 10.1006/jsre.1999.5754. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Marsden PA. Nitric oxide synthasis: biochemical and molecular regulation. Curr Opin Nephrol Hypertens. 1995;4:12–22. doi: 10.1097/00041552-199501000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Cheng C, Li X, Gao S, Niu S, Chen M, Qin J, Guo Z, Zhao J, Shen A. Expression of CAPON after spinal cord injury in rats. J Mol Neurosci. 2008;34:109–119. doi: 10.1007/s12031-007-9019-5. [DOI] [PubMed] [Google Scholar]
- 35.Hara MR, Snyder SH. Cell Signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–141. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- 36.Kowaltowski AJ, Vercesi AE, Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome C release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–910. doi: 10.1038/sj.cdd.4400722. [DOI] [PubMed] [Google Scholar]
- 37.Mishra OP, Zubrow AB, Ashraf QM, Delivoria-Papadopoulos M. Effect of nitric oxide synthase inhibition during post-hypoxic reoxygenation on Bax and Bcl-2 protein expression and DNA fragmentation in neuronal nuclei of newborn piglets. Brain Res. 2006;1101:20–28. doi: 10.1016/j.brainres.2006.05.021. [DOI] [PubMed] [Google Scholar]