

Abstract
Pediatric soft tissue sarcomas are a group of malignant neoplasms arising within embryonic mesenchymal tissues during the process of differentiation into muscle, fascia and fat. The tumors have a biphasic peak for age of incidence. Rhabdomyosarcoma (RMS) is diagnosed more frequently in younger children, whereas adult-type non-RMS soft tissue sarcoma is predominately observed in adolescents. The latter group comprises a variety of rare tumors for which diagnosis can be difficult and typically requires special studies, including immunohistochemistry and molecular genetic analysis. Current management for the majority of pediatric sarcomas is based on the data from large multi-institutional trials, which has led to great improvements in outcomes over recent decades. Although surgery remains the mainstay of treatment, the curative aim cannot be achieved without adjuvant treatment. Pre-treatment staging and risk classification are of prime importance in selecting an effective treatment protocol. Tumor resectability, the response to induction chemotherapy, and radiation generally determine the risk-group, and these factors are functions of tumor site, size and biology. Surgery provides the best choice of local control of small resectable tumors in a favorable site. Radiation therapy is added when surgery leaves residual disease or there is evidence of regional spread. Chemotherapy aims to reduce the risk of relapse and improve overall survival. In addition, upfront chemotherapy reduces the aggressiveness of the required surgery and helps preserve organ function in a number of cases. Long-term survival in low-risk sarcomas is feasible, and the intensity of treatment can be reduced. In high-risk sarcoma, current research is allowing more effective disease control.
Keywords: Pediatric tumor, Rhabdomyosarcoma, Soft tissue sarcoma, Non-rhabdomyosarcoma pediatric soft tissue tumor
Core tip: The manuscript describes current management of pediatric soft tissue sarcomas, a large group of rare tumors in pediatric age group. The group has two main categories; rhabdomyosarcoma (RMS) and non-RMS pediatric soft tissue tumors. Treatment of these tumors is in multidisciplinary fashion comprising of surgery, chemotherapy and radiation therapy. Decision making in management protocol for each patient is based on the risk determined by various clinical and pathological parameters. For cases with low-risk, surgical removal is usually adequate when adjuvant chemoradiation are proven helpful in cases with significant risk of recurrence. The overall survival in these tumors has become brighter in the recent decades.
INTRODUCTION
Pediatric soft tissue sarcomas are part of a heterogeneous group of tumors originating from embryonic mesodermal tissues during the process of differentiation into various mesenchymal tissue components of the human body. These tumors constitute 6% to 8% of all cancers in children less than 15 years of age[1-5]. Age-standardized incidence rates in Western countries are slightly increased compared with Asian countries[5]. Of all soft tissue sarcomas in this age group, approximately 50% to 60% are rhabdomyosarcoma (RMS), whereas the remainder are non-RMS soft tissue sarcomas (NRSTS), a designation that includes a variety of rarer soft tissue tumors including fibrosarcomas, synovial sarcomas, the extraosseous Ewing’s family of tumors, malignant peripheral nerve sheath tumors (MPNSTs) and inflammatory myofibroblastic tumors (IMT)[6,7]. According to the International Classification of Childhood Cancers, version 3, Kaposi sarcoma is also categorized as a NRSTS tumors[8]. Approximately two-thirds of RMSs are diagnosed before 6 years of age, and the incidence decreases with age[7,9]. In contrast, NRSTSs occur in older children, increasing in incidence throughout adolescent years[6]. In African countries wherein the human immunodeficiency virus is endemic, an exceptionally increased incidence of Kaposi sarcoma has been reported[2].
Although most soft tissue sarcomas occur sporadically, these lesions are associated with cancer predisposition syndrome in some patients, (e.g., Li-Fraumeni syndrome, which is linked to p53 germline mutations). Neurofibrosarcomas typically develop in individuals affected with neurofibromatosis type 1, an autosomal dominant disorder caused by mutations in the neurofibromatosis 1 gene (NF1). Individuals harboring germline mutations of NF1 are also prone to the development of embryonal RMS[10]. At the somatic level, specific chromosomal translocations and an expression of chimeric transcription factors are molecular signatures in a number of pediatric sarcomas. In RMS, PAX-FOXO1 fusion is a characteristic of the unfavorable histology or alveolar RMS. Such specific molecular patterns help differentiate sarcoma subtypes in which accurate pathological diagnosis may be difficult at a histopathological level.
The outcomes of pediatric soft tissue sarcomas have improved significantly during the past 3 decades[11]. The prognosis of pediatric soft tissue sarcoma, particularly RMS in younger children, is far better than that for sarcomas in adults. With modern evidence-based medicine, a multidisciplinary therapeutic approach not only increases survival rates but also provides a better chance to preserve the affected organ, particularly in the extremities and genitourinary organs. This article reviews current management practices for pediatric soft tissue sarcomas, with an emphasis on RMS and some soft tissue tumors that are more commonly found in the pediatric age group.
CLINICAL PRESENTATION AND DIAGNOSIS OF PEDIATRIC SOFT TISSUE SARCOMA
As soft tissue sarcomas are derived from primitive mesenchymal cells during their development into various mature mesenchymal tissue types (including muscle, fascia and fat), these tumors can be located in any part of the human body. The most common sites of primary RMSs are the head and neck, the genitourinary system and the limbs. The classic presentation is a growing lump that may or may not affect the function of nearby organs. RMS in some organ systems may cause specific symptoms. For example, frequent urination can be an initial presentation of an RMS that arises within the urinary bladder. Obstructive jaundice is one manifestation of bile duct RMS. Multiple plexiform neurofibrosarcomas can be benign tumors that follow neurofibrosarcoma in an individual with neurofibromatosis. Localization of the tumor site and its relation to the surrounding organs is typically accomplished by an imaging study, preferably magnetic resonance imaging (MRI) and/or computerized tomography (CT)[12]. From a surgical standpoint, the location, proximity to vascular structures, and potential morbidity caused by surgical resection determine the “resectability” of a sarcoma. To date, no serum markers are available for the diagnosis of soft tissue sarcomas. Image-guided core needle biopsy typically, but not always, provides a definitive diagnosis[13]. During a biopsy, extra tissue can be collected for further studies, i.e., electron microscopy and molecular diagnosis. Repeat biopsy using an open technique is performed when a histopathological diagnosis cannot be made upon examining a small strip of tissue obtained from a needle coring-out. Suspected lymph node metastasis should be confirmed by histopathology, particularly in sarcoma of the limbs and the paratesticular area.
Pre-treatment clinical staging aims to categorize the disease according to the tumor site, size, local invasion, regional lymph node involvement and distant metastasis. The metastatic work-up includes bone marrow aspiration/biopsy, bone scintigraphy, and axial imaging studies of the brain, lung and liver (CT or MRI). A spinal tap for cerebrospinal fluid is indicated in cases of suspected parameningeal tumor. A recent systematic review suggested the potential benefit of a functional imaging study, such as positron emission tomography (PET-CT), for increasing the accuracy of pretreatment staging, particularly in the evaluation of nodal status and distant metastasis[14,15]. Sentinel lymph node biopsy using radio-tracer exhibits feasibility and good concordance with PET-CT results in pediatric soft tissue sarcomas[16,17].
MULTIDISCIPLINARY MANAGEMENT OF PEDIATRIC SOFT TISSUE SARCOMA
RMS
RMS is a malignant mesenchymal tumor originating from immature striated muscle. Approximately 40% of RMSs occur in the head and neck region, 20% occur at genitourinary sites, 20% in the extremities, and 20% in other locations[9,12] (Figure 1). On hematoxylin and eosin histology, the tumor is characterized by the presence of spindle-shaped or small round-cell rhabdomyofibroblasts with eosinophilic cytoplasm. Cross-striations can be observed in some cases with relatively high tumor differentiation. Immunohistochemical studies that support the diagnosis of RMS include actin, desmin, myoglobin, myogenin and Myo-D. Pediatric RMS cases are generally categorized into 2 types: embryonal RMS (80%) and alveolar RMS (15%-20%)[12]. The botryoid subtype is a variant of embryonal RMS commonly located in the genitourinary tract, vagina, and biliary and nasopharyngeal sites. The spindle cell subtype is another subtype of embryonal RMS found in paratesticular locations. Alveolar RMS is observed in older children and generally has a more unfavorable histology.
Figure 1.
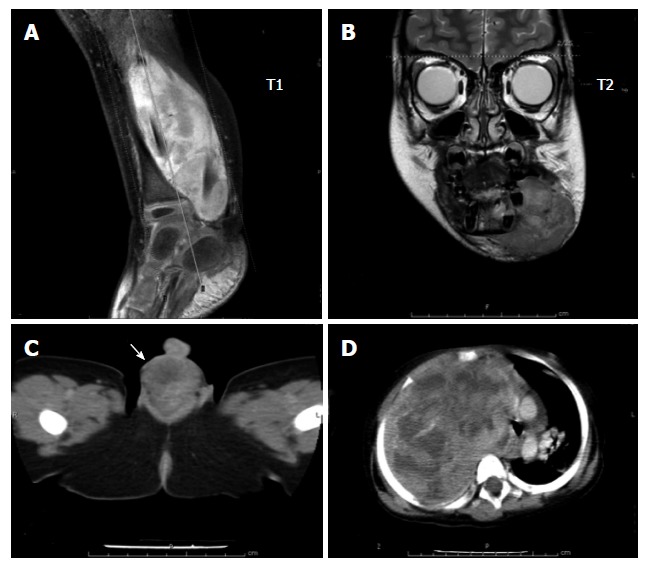
Radiographic images of common rhabdomyosarcoma. A: Extremity; B: Head and neck; C: Genitourinary (paratesticular); D: Axial (intraabdominal).
After the diagnosis is made, pre-treatment staging is performed according to a standard classification, such as the InterGroup Rhabdomyosarcoma Pretreatment Staging Classification (Table 1). The value of pretreatment staging involves determining disease prognosis. In addition to the stage, completeness of tumor removal defines the “clinical group” of RMS. Management plans for RMS can be divided into local control and systemic therapy and generally rely on risk categorization as determined by the Intergroup Rhabdomyosarcoma Studies (IRS) stage with the clinical group (Table 2). Surgery therefore has an integral role in the initial stages of decision-making on multimodality treatment in pediatric RMS.
Table 1.
Intergroup rhabdomyosarcoma study pretreatment staging and clinical grouping classification
| Stage | Sites | T1 | Size2 | N3 | M4 |
| 1 | Orbit, head and neck (excluding parameningeal), genitourinary tract (non-bladder, non-prostate), biliary tract | T1 or T2 | a or b | N0 or N1 or Nx | M0 |
| 2 | Bladder.Prostate, extremity, parameningeal, others (trunk, retroperitoneum, etc.) | T1 or T2 | a | N0 or Nx | M0 |
| 3 | Bladder.Prostate, extremity, parameningeal, others (trunk, retroperitoneum, etc.) | T1 or T2 | a | N1 | M0 |
| b | N0 or N1 or Nx | M0 | |||
| 4 | Any sites | T1 or T2 | a or b | N0 or N1 | M1 |
| Clinical group | Description | ||||
| I | Localized disease, completely resected | ||||
| II | Grossly resected tumor with evidence of regional spread | ||||
| IIA: Grossly resected tumor with microscopic residual disease | |||||
| IIB: Involved regional nodes completely resected with no microscopic residual disease | |||||
| IIC: Involved regional nodes grossly resected with evidence of microscopic residual disease | |||||
| III | Incomplete resection with gross residual disease after biopsy or after gross or major resection of the primary tumor | ||||
| IV | Distant metastatic disease present at diagnosis | ||||
T: Tumor; T1: Confined to the anatomic origin; T2: Extension and/or fixation to surrounding tissue;
Size, a: ≤ 5 cm; b: > 5 cm;
N: Regional nodes; N0: Regional nodes not clinically involved; Nx: Clinical status of regional nodes unknown; N1: Regional nodes clinically involved;
M: Metastasis; M0: No distant metastasis; M1: Distant metastasis present (includes positive cytology in pleural, peritoneal or cerebrospinal fluid).
Table 2.
Risk group stratification for rhabdomyosarcoma according to the International Rhabdomyosarcoma Study
| Risk group | Histology | Pretreatment stage | Clinical group |
| Low (subset 1) | Embryonal | 1 | I, II |
| 1 | III (orbit) | ||
| 2 | I, II | ||
| Low (subset 2) | 1 | III (non-orbit) | |
| 3 | I, II | ||
| Intermediate | Embryonal | 2, 3 | III |
| Alveolar | 1, 2, 3 | I, II, III | |
| High | Any | 4 | IV |
Local control
Surgery has been the most effective method to eliminate pathology. Surgery should be conducted in a manner that maintains function and cosmesis. In primary surgery, the extent of the initial surgery is generally subject to the judgment of the surgical team. In principle, resectability means that a tumor and its tumor-free surrounding tissue can be removed without operative risk or unacceptable post-operative morbidity. In the cases in which an excisional biopsy is performed without awareness of an adequate surgical margin, re-excision of the tumor bed should be considered[18,19]. In general, factors determining resectability include anatomical characters, such as site, size and vital structure involvement. However, in cases in which primary definitive surgery is not likely to provide complete resection without significant morbidity, delayed primary resection after upfront chemotherapy should be considered with an aim for organ salvage without compromising the long-term survival outcome. When delayed primary resection after neoadjuvant treatment is planned, compliance should also be considered. Intractable symptoms from the tumor and psychosocial factors may impact therapeutic compliance. Symptom control surgery during induction therapy (i.e., temporary urinary diversion in urinary bladder RMS) might be indicated[20]. Radical surgery is indicated in patients who are unable to tolerate intensive chemoradiation.
A recent multi-institutional data review demonstrated that approximately 90% of clinical group III embryonal RMS patients experienced a volume reduction of 33% or greater after induction chemotherapy[21]. Although the effect of the chemotherapeutic response on event-free survival (EFS) remains unclear, the study found that cases with at least a partial response experienced significantly enhanced overall survival (OS), particularly in head and neck RMS[21]. These results were consistent with a recent report using functional imaging tool 2-fluoro-2-deoxy-d-glucose-positron emission topography from the Memorial Sloan Kettering Cancer Center suggesting that the response after induction chemotherapy significantly predicted both EFS and OS[22]. An earlier report from the Intergroup Rhabdomyosarcoma Study IV (IRS-IV) revealed an 81% response to chemotherapy in group III RMS cases with no significant difference in the response rate between embryonal and alveolar RMS, and the size of the initial tumor had no influence on the response[23]. According to both studies, parameningeal RMS appeared to have a poorer response rate even when chemotherapy was administered with radiation[24]. Although the number of cases was lower, genitourinary tract sites (except bladder and prostate) exhibited better response rates[21]. The delayed primary resection strategy has reduced the extent of surgery in pelvic RMS. Pelvic exenteration, a historical standard in bladder and vaginal RMS, is rarely practiced today.
Second-look exploration aims to confirm the clinical/radiological response and to achieve oncologic resection when possible. Imaging evaluation may underestimate the degree of the response. According to IRS-III data, 46% of patients who achieved partial remission were found to be in complete remission at surgical exploration, and an additional 28% could be converted to complete remission. In addition, 30% of patients who had clinically stable disease after induction chemotherapy exhibited pathological complete remission, and an additional 43% could be converted to complete remission[23]. To achieve oncologic resection, radical organ removal must be performed in some situations. In urinary bladder RMS that arises at the base of the bladder or prostate, a partial cystectomy is not sufficient given the high risk of local failure. Total cystectomy and urologic conduit is a surgical option that potentially leads to long-term, disease-free survival with an acceptable quality of life. Bladder-preserving surgery is reserved for cases with a good response to induction chemoradiation therapy. In addition, the tumor location must allow a 2- to 3-cm tumor-free margin, and at least two-thirds of the bladder must be retained[20,25]. In vaginal RMS, residual tumor after chemotherapy is an indication for total hysterectomy with gonadal preservation[26]. Pancreaticoduodenectomy is the operation of choice in cases in which the RMS involves the distal common bile duct[27].
Lymph node management is of prime importance in pre-treatment staging and clinical grouping of RMS, both of which determine the risk category. Radical lymph node dissection does not impact outcome. Enlarged nodes detected clinically or by radiologic evidence should be excised for histopathological examination (Figure 2). Regardless of radiologic evidence, lymph node sampling is indicated in extremity RMS and for children older than 10 years with paratesticular tumor[9,11,28]. When an adjacent node is positive, more distant nodes should be searched for and biopsied. To reduce the morbidity caused by extensive lymph node sampling, the concept of sentinel lymph node sampling, which is the current standard in melanoma and breast cancer, has also been adapted for pediatric soft tissue sarcoma. Trials in pediatric sarcomas had relatively small numbers in each series[16,17,29]. Most studies used the lymphoscintigraphy technique and reported that the technique was feasible in pediatric sarcomas; however, specific data regarding identification rates and false negative rates in RMS remain inconclusive.
Figure 2.
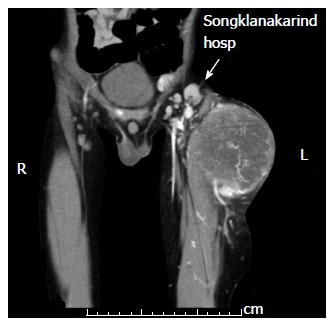
Inguinal lymph node enlargement in a case of extremity rhabdomyosarcoma.
Radiation therapy is unnecessary for embryonal RMS in clinical group I (completely resected) when it provides better failure-free survival in alveolar RMS. Radiation enhances local control in cases with residual disease after definitive surgery, positive locoregional lymph nodes and unresectable RMS after tumor biopsy. Radiation doses to microscopic residual tumors (total 36 Gy) are typically less than those for gross residual or primary unresectable tumors (50.4 Gy)[11]. Orbital tumors are an exception as their clinical group III requires 45 Gy. Data from the German trial CWS91 indicated that hyperfractionated accelerated radiotherapy may reduce the total radiation dose in RMS (32 Gy in low-risk and 48 Gy in high-risk patients) without compromising treatment outcomes[30]. Alternative radiation therapy techniques, such as intensity modulated radiation therapy, brachytherapy, and proton beam therapy, are used in some centers with the aim of reducing locoregional side effects[31].
Systemic therapy
Chemotherapy is an essential component of the multimodality treatment of RMS. The standard regimen in non-metastatic RMS is a combination of vincristine, actinomycin-D and cyclophosphamide (VAC)[32]. Omitting cyclophosphamide from the regimen has been attempted in low-risk RMS cases to reduce the cumulative dose of cyclophosphamide (IRS-D9602 protocol). Although VA produced an excellent outcome in a subset of low-risk RMS cases, including group I-II, stage 1-2 and group III orbital tumor (subset 1, Table 2), the data suggested that cyclophosphamide should be retained with vincristine and actinomycin-D in the other subset of low-risk RMS cases (subset 2: group I-II, stage 3 and group III, stage I except orbital tumor) because the failure-free survival was poorer than that of comparable patients in the IRS-IV study who received a triple drug regimen[33]. IRS-IV data also demonstrated that substitution of cyclophosphamide with ifosfamide (VAI) or substitution of actinomycin-D/cyclophosphamide with ifosfamide and etoposide did not improve the failure-free survival in non-metastatic RMS[34]. A subsequent study from the Children's Oncology Group (COG), directed toward a shorter duration of VA and dose reduction of cyclophosphamide (ARST0331) in low-risk RMS patients, was recently published. Although the study reported an increased incidence of local failure with use of the shorter therapy, the study recommended its use in low-risk RMS cases given the reduced toxicity[35]. For intermediate- and high-risk patients, successive COG trials have attempted to improve the survival outcome by incorporating novel agents, such as doxorubicin, ifosfamide and etoposide (VDC/IE), and irenotecan (VAC/VI), with the aim of reducing the cumulative cyclophosphamide dose[36]. Various molecular targeting drugs are being explored in high-risk RMS cases, including vascular endothelial growth factors (bevacizumab), mTOR (temsirolimus) and IGF-1R (temozolomide). A phase II trial of temozolomide has demonstrated its safety and feasibility; however, the preliminary response rate was not impressive[37]. For metastatic RMS, another study found that the incorporation of VDC-IE or VI with VAC therapy resulted in improved outcomes in embryonal RMS[38].
Outcome of current multimodality management in RMS
Since the establishment of the International Rhabdomyosarcoma Study Group in 1972 (currently the Cooperative Soft Tissue Sarcoma Study Group), survival of pediatric RMS patients has been steadily improving. Before the era of multimodality treatment, surgery alone resulted in survival rates less than 20% (11). With the new available treatments, the five-year OS increased from 55% in IRS-I to 63% in IRS-II and to 71% in IRS-III and IRS-IV. Data from IRS-II to IRS-IV revealed an 88% 3-year failure-free survival in low risk embryonal RMS. Intermediate-risk embryonal RMS had a 4-year failure-free survival rate of approximately 68% to 78%; however, survival in high-risk patients remains poor at less than 25%[39].
NON-RMS SOFT TISSUE SARCOMAS
NRSTSs are a heterogeneous group of rare mesenchymal tumors that exhibit a wide variety of histopathologies and biologies. The majority of NRSTSs occur more frequently in adult patients, and the prognosis is generally poorer than for pediatric sarcomas. Given their heterogeneity, ambiguity in pathological diagnosis is common, and care should be taken when obtaining tissue samples[40]. A multidisciplinary conference before the initiation of treatment for any individual case allows the team to arrive at a consensus and understand the role of each discipline in the treatment process. Surgery has a primary role in the treatment of resectable NRSTSs, whereas adjuvant treatment relies on a Children’s Oncology Group risk stratification guideline[41] (Table 3). Basically, radiation therapy is administered to patients whose resection margins are close to the tumor (except for very low-risk tumors). Chemotherapy provides a poorer response than pediatric RMS and is advocated in select types of NRSTS. Ifosfamide and doxorubicin are backbones recommended as post-operative adjuvant therapy for localized resectable STSs[42,43]. A recent systematic review found that autologous hematopoietic stem cell transplantation following high-dose chemotherapy in locally advanced or metastatic NRSTS did not result in better OS than standard-dose chemotherapy[44].
Table 3.
Risk stratification in nonrhabdomyosarcoma soft tissue sarcoma and treatment proposal according to the Children's Oncology Group (NCT00346164)
| Risk group |
Factors |
Proposed treatment | |||
| Grade | Size | Stage | Initial resectability | ||
| Low | Low | Any | Nonmetastatic | Gross resection | Observation |
| High | < 5 cm | Nonmetastatic | Without microscopic margins | Observation | |
| High | < 5 cm | Nonmetastatic | With microscopic margin | Adjuvant radiation therapy | |
| Intermediate | High | > 5 cm | Nonmetastatic | Gross resection | Adjuvant chemotherapy and radiation therapy |
| High | > 5 cm | Nonmetastatic | Unresected | Neoadjuvant chemoradiotherapy, surgery, adjuvant chemotherapy with or without radiation therapy | |
| High | Low | Any | Metastatic | Gross resection | Observation |
| High | Any | Metastatic | Gross resection | Adjuvant chemotherapy and radiation therapy | |
| High | Any | Metastatic | Unresected | Neoadjuvant chemoradiotherapy, surgery, adjuvant chemotherapy with or without radiation therapy | |
Extraosseous Ewing’s sarcoma family of tumors
Extraosseous primitive neuroectodermal tumors, namely, Ewing’s sarcoma and Adkin tumor of the chest wall, are grouped together as the Ewing’s family of tumors because they share a chromosomal translocation, t(11;22)(q24;q12), leading to a chimeric fusion, EWSR1-FLI1[45]. Extraosseous Ewing’s sarcoma presents predominately in the second decade of life. The tumor is comprises 15% to 20% of all Ewing’s sarcomas[46,47]. The tumor can occur anywhere in the body but commonly presents on the extremities, chest and pelvis. Histologically, the tumor belongs to the small round blue cell group and demonstrates positive immunoreactivity to the surface glycoprotein CD99. Poor prognostic indicators include axial site tumors, particularly in the pelvic region; large tumor size; late stage; poor response to induction chemotherapy; advanced age; and high serum lactate dehydrogenase levels[46-49]. The definitive treatment for extraosseous EFSTs is surgical removal. Complete resection is the best option for cure, and the likelihood of achieving negative surgical margins is increased when induction chemotherapy is administered[50]. Although these tumors are relatively sensitive to radiation, radiation is reserved for cases with positive surgical margins or incompletely resected tumor because late effects of radiation, such as a second malignancy, are of concern. Post-operative chemotherapy aims to improve OS and reduce the likelihood of local recurrence. The therapeutic regimen in extraosseous EFST follows that used in either NRSTS or Ewing’s sarcoma of the bone and typically comprises ifosfamide/etoposide with or without carboplatin (ICE) alternated with a combination of vincristine, doxorubicin and cyclophosphamide (VDC)[47]. The results from the EICESS-92 study and the successive trial Euro-Ewing-99-R1 from the European InterGroup Cooperative Ewing’s Sarcoma Study concluded that ifosfamide can be substituted with cyclophosphamide in the consolidation phase in standard risk EFST (localized tumor with either good histological response after induction chemotherapy, small tumor resected at diagnosis, or receiving radiotherapy alone as a local treatment)[48,51]. A report from the French Society of Pediatric Oncology (SFOP-EW93) suggested that induction with cyclophosphamide and doxorubicin followed by histopathological response-based chemotherapy (VAC or VAC/VIE or IE + high dose busulfan/melphalan) provided superior outcomes to those of an ifosfamide-based regimen (VAI) for all cases[52]. Other studies reported a five-year OS of between 60% and 70% in non-metastatic extraosseous EFSTs[46,52,53], whereas another study reported that metastatic EFST exhibited a 5-year OS of approximately 25%[53].
MPNST
MPNSTs, malignant schwannomas, neurofibrosarcomas and neurogenic sarcomas account for approximately 6% of all NRSTSs[54], and approximately half of these cases are associated with neurofibromatosis type 1 syndrome[55]. An individual with the NF1 mutation has a cumulative 8% to 13% lifetime risk of developing MPNST[56]. MPNST develops within benign neurofibromas in NF1 patients[57]. In the pediatric age group, the incidence increases with age, with more than 80% of cases diagnosed at the age 10 years or older[58]. Among pediatric NRSTSs, the tumor has the worst prognosis, with a 5-year OS of 43% to 59%[59]. Complete surgical removal is the only chance for cure. Unfortunately, a number of MPNSTs involve the nerve root, preventing complete removal (Figure 3). Radiation therapy is recommended in cases with residual tumor after surgery; however, no evidence indicates that this improves survival[60]. Studies have reported that adjuvant chemotherapy exhibits only minimal benefit[58,61,62].
Figure 3.
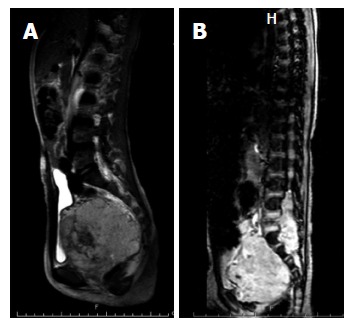
Magnetic resonance imaging T2: Providing a comparison between growth patterns. A: Pelvic RMS; B: MPNST. Although RMS is a locally advanced tumor, a thin surgical plane typically exists between the tumor and the adjacent bone when the MPNST involves the dural space and nerve root. RMS: Rhabdomyosarcoma; MPNST: Malignant peripheral nerve sheath tumor.
Synovial sarcoma
Synovial sarcoma (SS) is an aggressive spindle cell tumor that accounts for approximately 10% of all STSs[63]. Although the tumor is principally located in the lower extremities, primary SS at other sites (including the head and neck, hands, retroperitoneum, digestive system and mediastinum) have been reported[64-66]. Histologically, SS contains spindle cells with a varying degree of epithelial differentiation[67]. On immunohistochemical study, SS is marked with both mesenchymal and epithelial markers. The cytogenetic signature of SS is a reciprocal translocation t(X;18) (p11.2;q11.2) that leads to a chimeric fusion between SS18 from chromosome 18 and one of the SSXs (SSX1, SSX2 or SSX4) from chromosome X. The SS18-SSX2 fusion protein activates canonical Wnt/beta-catenin signaling, which suggests a future therapeutic target in a subset of SS[68,69]. The current management of SS is based on risk categorization, and risk determinants include the clinical group (as in RMS), size (5 cm) and sites[70]. Low-risk tumors include group I SS and are less than 5 cm in size. Axial site tumors (head and neck, trunk, lungs and pleura) are considered high risk[71]. According to the European Pediatric Soft Tissue Sarcoma Study Group Trial (EpSSG NRSTS2005), low-risk SSs are best treated with surgery alone, with 91.7% experiencing 3-year EFS and 100% OS[71]. In that study, the surgical strategy recommended in most low-risk cases was conservative surgery. Survival in intermediate-risk SSs (group I, size > 5 cm and group II) after surgery followed by chemotherapy (ifosfamide and doxorubicin) with or without radiation is comparable with that of the low-risk group. Chemotherapy is the mainstay treatment in high-risk (group III or axial SS) patients. The chemotherapy response rate in group III SS was 55%, and OS was 74%[71].
Congenital infantile fibrosarcoma
Unlike tumors in the adult-type NRSTS group that are typically found in teenagers and adolescents, congenital infantile fibrosarcoma (CIF) can be noted during the first month of life and is often misdiagnosed as a hemangioma or vascular malformation. A rapid growth rate and ulceration are clinical clues necessitating a biopsy[72]. Histologically, CIF is densely packed with spindle cells arranged in bundles and fascicles. Tumor cells typically exhibit positive immunoreactivity with the mesenchymal marker vimentin but are negative for desmin and S100 protein. A chromosomal translocation t(12;15)(p13;q25), which leads to a fusion ETV6-NTRK3, has been reported. The tumor is locally aggressive, and distant metastasis is rarely reported. Destruction of adjacent bony structures can be observed (Figure 4). Surgical removal of the lesion is the recommended primary treatment. Adjuvant treatment is generally unnecessary except when the mass is very large and involves vital structures. In such instances, neoadjuvant chemotherapy may help down-size the tumor[73,74]. Prognostic factors include the site and extent of the lesion at the diagnosis. Extremity IF has a more favorable outcome than do axial tumors. In addition, pediatric CIF has a better outcome than adult fibrosarcoma. The five-year OS is approximately 90%[75,76].
Figure 4.
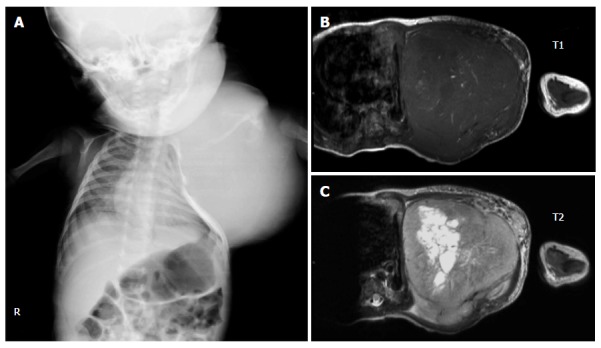
Plain radiographic and magnetic resonance images demonstrating deformity of the left chest wall caused by a congenital infantile sarcoma (A, B and C).
Desmoplastic small round cell tumors
Desmoplastic small round cell tumors (DSRCT) is a rare, highly aggressive mesenchymal tumor originating on the peritoneal surface typically in an adolescent[77]. The tumor can also be found at other sites, such as the head and neck, pleura, kidneys, ovaries and testes[78-81] and was first described in 1989 in a pathological case report by Gerald and Rosai[82]. Histologically, DSRCT exhibits small round cells arranged in nests within abundant desmoplastic stroma[77]. Central necrosis and trabecular or indian fire arrangements are also observed[83]. The tumor expresses polyphenotypic differentiation with co-expression of epithelial, mesenchymal and neuronal markers[77]. In addition, nuclear staining of the WT1 protein has been reported[83]. The tumor is highly aggressive, and approximately 60% of patients die of the disease within 2 years[84]. Complete resection is not possible in the majority of cases. Surgical debulking of the primary tumor followed by radiation therapy is recommended[85,86]. The tumor appears to respond to multi-agent chemotherapy consisting of cyclophosphamide, doxorubicin, vincristine, ifosfamide and etoposide; however, recurrent disease is common[85,87]. The use of alternative therapies, including molecular targeting therapy and intraperitoneal infusion of chemotherapy, is reported infrequently[88-90]. In one study, the 3-year OS was reported at 44%, with a 5-year OS of 15%[85].
IMT
IMT (IMT, also known as inflammatory pseudotumor or plasma cell granuloma) is a rare benign tumor with recurrence potential that most often occurs in children and young adults. The lung is the most common site of IMT. Other reported sites include the urinary bladder, intestine and mesentery, spleen, liver and kidney[91-94]. The etiology of IMT may include certain infections, such as Epstein Barr virus. Whether the tumor is a true neoplasm or an inflammatory response remains controversial. However, recurrence after surgery is common and malignant transformation has been reported[95,96]. Studies have demonstrated that a number of IMT involve fusion between the AKT gene in chromosome 2 (2q23) and various fusion partners, including IMT, TPM3, TPM4, CLTC, CARS, RANBP2, ATIC, SEC 31L1 and PPFBP1[97-103]. CT typically reveals a coin lesion that is difficult to differentiate from other causes of similar lesions. The diagnosis is typically made by tissue biopsy that often exhibit spindle-shaped myofibroblast-like cells and chronic inflammation comprising plasma cells, lymphocytes and histiocytes[104]. Surgical resection is the only treatment option. Radiation and chemotherapy have no role in IMT (Figure 5).
Figure 5.
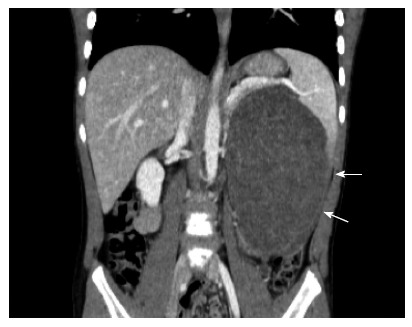
Computerized tomography image of a case of splenic inflammatory myofibroblastic tumor.
STSs in the pediatric age group are a heterogeneous group of rare mesenchymal tumors. Survival outcome in pediatric STS has improved since cooperative studies were initiated by various international organizations, particularly the International Rhabdomyosarcoma Study Group. Treatment of these tumors relies on knowledge of their natural history and tumor biology as this information is used to categorize STSs according to their risk. Although surgery has been the main treatment in localized low-risk tumors, good outcomes are not achieved without adjuvant radiation and chemotherapy. Future studies in the treatment of STS are directed toward the use of molecular diagnosis as an integral part of tumor classification. While novel modalities for the treatment of advanced stage tumors are under investigation, trials should be conducted on the reduction of treatment intensity in low-risk patients.
ACKNOWLEDGMENTS
The Anandamahidol Foundation, Thailand provided support to Sangkhathat S for this work. The inclusion of clinical and radiological pictures was approved by the Institution Review Board of the Faculty of Medicine, Prince of Songkla University. The author acknowledges the help of Dave Patterson in English language editing for the manuscript.
Footnotes
Supported by The Anandamahidol Foundation.
Conflict-of-interest statement: None declared.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 2, 2015
First decision: July 3, 2015
Article in press: September 30, 2015
P- Reviewer: Chen MK, Mostafa BE S- Editor: Tian YL L- Editor: A E- Editor: Jiao XK
References
- 1.Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev. 2010;36:277–285. doi: 10.1016/j.ctrv.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Hadley LG, Rouma BS, Saad-Eldin Y. Challenge of pediatric oncology in Africa. Semin Pediatr Surg. 2012;21:136–141. doi: 10.1053/j.sempedsurg.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001-2009. Pediatrics. 2014;134:e945–e955. doi: 10.1542/peds.2013-3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 5.Liu YL, Lo WC, Chiang CJ, Yang YW, Lu MY, Hsu WM, Ho WL, Li MJ, Miser JS, Lin DT, et al. Incidence of cancer in children aged 0-14 years in Taiwan, 1996-2010. Cancer Epidemiol. 2015;39:21–28. doi: 10.1016/j.canep.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Loeb DM, Thornton K, Shokek O. Pediatric soft tissue sarcomas. Surg Clin North Am. 2008;88:615–627, vii. doi: 10.1016/j.suc.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapoor G, Das K. Soft tissue sarcomas in children. Indian J Pediatr. 2012;79:936–942. doi: 10.1007/s12098-011-0560-4. [DOI] [PubMed] [Google Scholar]
- 8.Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International Classification of Childhood Cancer, third edition. Cancer. 2005;103:1457–1467. doi: 10.1002/cncr.20910. [DOI] [PubMed] [Google Scholar]
- 9.Dasgupta R, Rodeberg DA. Update on rhabdomyosarcoma. Semin Pediatr Surg. 2012;21:68–78. doi: 10.1053/j.sempedsurg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Crucis A, Richer W, Brugières L, Bergeron C, Marie-Cardine A, Stephan JL, Girard P, Corradini N, Munzer M, Lacour B, et al. Rhabdomyosarcomas in children with neurofibromatosis type I: A national historical cohort. Pediatr Blood Cancer. 2015;62:1733–1738. doi: 10.1002/pbc.25556. [DOI] [PubMed] [Google Scholar]
- 11.Paulino AC, Okcu MF. Rhabdomyosarcoma. Curr Probl Cancer. 2008;32:7–34. doi: 10.1016/j.currproblcancer.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Leaphart C, Rodeberg D. Pediatric surgical oncology: management of rhabdomyosarcoma. Surg Oncol. 2007;16:173–185. doi: 10.1016/j.suronc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Ilivitzki A, Abugazala M, Arkovitz M, Benbarak A, Postovsky S, Arad-Cohen N, Ben-Arush M. Ultrasound-guided core biopsy as the primary tool for tissue diagnosis in pediatric oncology. J Pediatr Hematol Oncol. 2014;36:333–336. doi: 10.1097/MPH.0b013e31827e4c4d. [DOI] [PubMed] [Google Scholar]
- 14.Federico SM, Spunt SL, Krasin MJ, Billup CA, Wu J, Shulkin B, Mandell G, McCarville MB. Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer. 2013;60:1128–1134. doi: 10.1002/pbc.24430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norman G, Fayter D, Lewis-Light K, McHugh K, Levine D, Phillips B. Mind the gap: extent of use of diffusion-weighted MRI in children with rhabdomyosarcoma. Pediatr Radiol. 2015;45:778–781. doi: 10.1007/s00247-014-3120-z. [DOI] [PubMed] [Google Scholar]
- 16.Alcorn KM, Deans KJ, Congeni A, Sulkowski JP, Bagatell R, Mattei P, Minneci PC. Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg. 2013;48:1903–1906. doi: 10.1016/j.jpedsurg.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 17.Dall’Igna P, De Corti F, Alaggio R, Cecchetto G. Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg. 2014;24:482–487. doi: 10.1055/s-0034-1396422. [DOI] [PubMed] [Google Scholar]
- 18.Cecchetto G, Guglielmi M, Inserra A, Zanetti I, Dall’Igna P, Gigante C, Carli M. Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int. 2001;17:532–534. doi: 10.1007/s003830100580. [DOI] [PubMed] [Google Scholar]
- 19.Chui CH, Spunt SL, Liu T, Pappo AS, Davidoff AM, Rao BN, Shochat SJ. Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg. 2002;37:1424–1429. doi: 10.1053/jpsu.2002.35405. [DOI] [PubMed] [Google Scholar]
- 20.Komasara L, Gołębiewski A, Anzelewicz S, Czauderna P. A review on surgical techniques and organ sparing procedures in bladder/prostate rhabdomyosarcoma. Eur J Pediatr Surg. 2014;24:467–473. doi: 10.1055/s-0034-1396424. [DOI] [PubMed] [Google Scholar]
- 21.Dantonello TM, Stark M, Timmermann B, Fuchs J, Selle B, Linderkamp C, Handgretinger R, Hagen R, Feuchtgruber S, Kube S, et al. Tumour volume reduction after neoadjuvant chemotherapy impacts outcome in localised embryonal rhabdomyosarcoma. Pediatr Blood Cancer. 2015;62:16–23. doi: 10.1002/pbc.25207. [DOI] [PubMed] [Google Scholar]
- 22.Casey DL, Wexler LH, Fox JJ, Dharmarajan KV, Schoder H, Price AN, Wolden SL. Predicting outcome in patients with rhabdomyosarcoma: role of [(18)f]fluorodeoxyglucose positron emission tomography. Int J Radiat Oncol Biol Phys. 2014;90:1136–1142. doi: 10.1016/j.ijrobp.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg AR, Anderson JR, Lyden E, Rodeberg DA, Wolden SL, Kao SC, Parham DM, Arndt C, Hawkins DS. Early response as assessed by anatomic imaging does not predict failure-free survival among patients with Group III rhabdomyosarcoma: a report from the Children’s Oncology Group. Eur J Cancer. 2014;50:816–823. doi: 10.1016/j.ejca.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladra MM, Mandeville HC, Niemierko A, Padera TP, Friedmann AM, MacDonald SM, Ebb D, Chen YL, Tarbell NJ, Yock TI. Local failure in parameningeal rhabdomyosarcoma correlates with poor response to induction chemotherapy. Int J Radiat Oncol Biol Phys. 2015;92:358–367. doi: 10.1016/j.ijrobp.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein R, Frees S, Schröder A, Russo A, Gutjahr P, Faber J, Thüroff JW. Radical surgery and different types of urinary diversion in patients with rhabdomyosarcoma of bladder or prostate--a single institution experience. J Pediatr Urol. 2013;9:932–939. doi: 10.1016/j.jpurol.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 26.van Sambeeck SJ, Martens SJ, Hundscheid T, Janssen EJ, Vos GD. Dutch paediatrician’s opinions about acute care for critically ill children in general hospitals. Eur J Pediatr. 2015;174:607–613. doi: 10.1007/s00431-014-2439-7. [DOI] [PubMed] [Google Scholar]
- 27.d’Ambrosio G, del Prete L, Grimaldi C, Bertocchini A, Lo Zupone C, Monti L, de Ville de Goyet J. Pancreaticoduodenectomy for malignancies in children. J Pediatr Surg. 2014;49:534–538. doi: 10.1016/j.jpedsurg.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Dang ND, Dang PT, Samuelian J, Paulino AC. Lymph node management in patients with paratesticular rhabdomyosarcoma: a population-based analysis. Cancer. 2013;119:3228–3233. doi: 10.1002/cncr.28198. [DOI] [PubMed] [Google Scholar]
- 29.Gow KW, Rapkin LB, Olson TA, Durham MM, Wyly B, Shehata BM. Sentinel lymph node biopsy in the pediatric population. J Pediatr Surg. 2008;43:2193–2198. doi: 10.1016/j.jpedsurg.2008.08.063. [DOI] [PubMed] [Google Scholar]
- 30.Dantonello TM, Int-Veen C, Harms D, Leuschner I, Schmidt BF, Herbst M, Juergens H, Scheel-Walter HG, Bielack SS, Klingebiel T, et al. Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol. 2009;27:1446–1455. doi: 10.1200/JCO.2007.15.0466. [DOI] [PubMed] [Google Scholar]
- 31.Egas-Bejar D, Huh WW. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc Health Med Ther. 2014;5:115–125. doi: 10.2147/AHMT.S44582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruymann FB. The development of VAC chemotherapy in rhabdomyosarcoma: what does one do for an encore? Curr Oncol Rep. 2003;5:505–509. doi: 10.1007/s11912-003-0012-z. [DOI] [PubMed] [Google Scholar]
- 33.Raney RB, Walterhouse DO, Meza JL, Andrassy RJ, Breneman JC, Crist WM, Maurer HM, Meyer WH, Parham DM, Anderson JR. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2011;29:1312–1318. doi: 10.1200/JCO.2010.30.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, Breneman J, Qualman SJ, Wiener E, Wharam M, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19:3091–3102. doi: 10.1200/JCO.2001.19.12.3091. [DOI] [PubMed] [Google Scholar]
- 35.Walterhouse DO, Pappo AS, Meza JL, Breneman JC, Hayes-Jordan AA, Parham DM, Cripe TP, Anderson JR, Meyer WH, Hawkins DS. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2014;32:3547–3552. doi: 10.1200/JCO.2014.55.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss A, Gill J, Goldberg J, Lagmay J, Spraker-Perlman H, Venkatramani R, Reed D. Advances in therapy for pediatric sarcomas. Curr Oncol Rep. 2014;16:395. doi: 10.1007/s11912-014-0395-z. [DOI] [PubMed] [Google Scholar]
- 37.Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, Ladanyi M, Grippo JF, Dall G, Staddon AP, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014;120:2448–2456. doi: 10.1002/cncr.28728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDowell HP, Foot AB, Ellershaw C, Machin D, Giraud C, Bergeron C. Outcomes in paediatric metastatic rhabdomyosarcoma: results of The International Society of Paediatric Oncology (SIOP) study MMT-98. Eur J Cancer. 2010;46:1588–1595. doi: 10.1016/j.ejca.2010.02.051. [DOI] [PubMed] [Google Scholar]
- 39.Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM, Qualman SJ, Wharam MD, Donaldson SS, Maurer HM, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003;21:78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 40.Magro G, Longo FR, Angelico G, Spadola S, Amore FF, Salvatorelli L. Immunohistochemistry as potential diagnostic pitfall in the most common solid tumors of children and adolescents. Acta Histochem. 2015;117:397–414. doi: 10.1016/j.acthis.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Waxweiler TV, Rusthoven CG, Proper MS, Cost CR, Cost NG, Donaldson N, Garrington T, Greffe BS, Heare T, Macy ME, et al. Non-Rhabdomyosarcoma Soft Tissue Sarcomas in Children: A Surveillance, Epidemiology, and End Results Analysis Validating COG Risk Stratifications. Int J Radiat Oncol Biol Phys. 2015;92:339–348. doi: 10.1016/j.ijrobp.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113:573–581. doi: 10.1002/cncr.23592. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka K, Mizusawa J, Fukuda H, Araki N, Chuman H, Takahashi M, Ozaki T, Hiruma T, Tsuchiya H, Morioka H, et al. Perioperative chemotherapy with ifosfamide and doxorubicin for high-grade soft tissue sarcomas in the extremities (JCOG0304) Jpn J Clin Oncol. 2015;45:555–561. doi: 10.1093/jjco/hyv042. [DOI] [PubMed] [Google Scholar]
- 44.Peinemann F, Labeit AM. Autologous haematopoietic stem cell transplantation following high-dose chemotherapy for non-rhabdomyosarcoma soft tissue sarcomas: a Cochrane systematic review. BMJ Open. 2014;4:e005033. doi: 10.1136/bmjopen-2014-005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 46.Biswas B, Shukla NK, Deo SV, Agarwala S, Sharma DN, Vishnubhatla S, Bakhshi S. Evaluation of outcome and prognostic factors in extraosseous Ewing sarcoma. Pediatr Blood Cancer. 2014;61:1925–1931. doi: 10.1002/pbc.25095. [DOI] [PubMed] [Google Scholar]
- 47.Brunetto AL, Castillo LA, Petrilli AS, Macedo CD, Boldrini E, Costa C, Almeida MT, Kirst D, Rodriguez-Galindo C, Pereira WV, et al. Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr Blood Cancer. 2015;62:1747–1753. doi: 10.1002/pbc.25562. [DOI] [PubMed] [Google Scholar]
- 48.Obata H, Ueda T, Kawai A, Ishii T, Ozaki T, Abe S, Tanaka K, Tsuchiya H, Matsumine A, Yabe H. Clinical outcome of patients with Ewing sarcoma family of tumors of bone in Japan: the Japanese Musculoskeletal Oncology Group cooperative study. Cancer. 2007;109:767–775. doi: 10.1002/cncr.22481. [DOI] [PubMed] [Google Scholar]
- 49.Duchman KR, Gao Y, Miller BJ. Prognostic factors for survival in patients with Ewing’s sarcoma using the surveillance, epidemiology, and end results (SEER) program database. Cancer Epidemiol. 2015;39:189–195. doi: 10.1016/j.canep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 50.Shamberger RC, LaQuaglia MP, Gebhardt MC, Neff JR, Tarbell NJ, Marcus KC, Sailer SL, Womer RB, Miser JS, Dickman PS, et al. Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg. 2003;238:563–567; discussion 567-568. doi: 10.1097/01.sla.0000089857.45191.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paulussen M, Craft AW, Lewis I, Hackshaw A, Douglas C, Dunst J, Schuck A, Winkelmann W, Köhler G, Poremba C, et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26:4385–4393. doi: 10.1200/JCO.2008.16.5720. [DOI] [PubMed] [Google Scholar]
- 52.Castex MP, Rubie H, Stevens MC, Escribano CC, de Gauzy JS, Gomez-Brouchet A, Rey A, Delattre O, Oberlin O. Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol. 2007;25:1176–1182. doi: 10.1200/JCO.2005.05.0559. [DOI] [PubMed] [Google Scholar]
- 53.Raney RB, Asmar L, Newton WA, Bagwell C, Breneman JC, Crist W, Gehan EA, Webber B, Wharam M, Wiener ES, et al. Ewing’s sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol. 1997;15:574–582. doi: 10.1200/JCO.1997.15.2.574. [DOI] [PubMed] [Google Scholar]
- 54.Spunt SL, Pappo AS. Childhood nonrhabdomyosarcoma soft tissue sarcomas are not adult-type tumors. J Clin Oncol. 2006;24:1958–1959; author reply 1959-1960. doi: 10.1200/JCO.2005.05.4957. [DOI] [PubMed] [Google Scholar]
- 55.Pourtsidis A, Doganis D, Baka M, Bouhoutsou D, Varvoutsi M, Synodinou M, Giamarelou P, Kosmidis H. Malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. Case Rep Oncol Med. 2014;2014:843749. doi: 10.1155/2014/843749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573–1577. [PubMed] [Google Scholar]
- 57.Schaefer IM, Fletcher CD. Malignant peripheral nerve sheath tumor (MPNST) arising in diffuse-type neurofibroma: clinicopathologic characterization in a series of 9 cases. Am J Surg Pathol. 2015;39:1234–1241. doi: 10.1097/PAS.0000000000000447. [DOI] [PubMed] [Google Scholar]
- 58.Bates JE, Peterson CR, Dhakal S, Giampoli EJ, Constine LS. Malignant peripheral nerve sheath tumors (MPNST): a SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr Blood Cancer. 2014;61:1955–1960. doi: 10.1002/pbc.25149. [DOI] [PubMed] [Google Scholar]
- 59.Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, Cecchetto G, Alaggio R, De Sio L, Koscielniak E, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23:8422–8430. doi: 10.1200/JCO.2005.01.4886. [DOI] [PubMed] [Google Scholar]
- 60.Kahn J, Gillespie A, Tsokos M, Ondos J, Dombi E, Camphausen K, Widemann BC, Kaushal A. Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front Oncol. 2014;4:324. doi: 10.3389/fonc.2014.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers PA, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 62.Milano GM, Cozza R, Ilari I, De Sio L, Boldrini R, Jenkner A, De Ioris M, Inserra A, Dominici C, Donfrancesco A. High histologic and overall response to dose intensification of ifosfamide, carboplatin, and etoposide with cyclophosphamide, doxorubicin, and vincristine in patients with high-risk Ewing sarcoma family tumors: the Bambino Gesù Children’s Hospital experience. Cancer. 2006;106:1838–1845. doi: 10.1002/cncr.21780. [DOI] [PubMed] [Google Scholar]
- 63.Merchant MS, Mackall CL. Current approach to pediatric soft tissue sarcomas. Oncologist. 2009;14:1139–1153. doi: 10.1634/theoncologist.2009-0160. [DOI] [PubMed] [Google Scholar]
- 64.Hu PA, Zhou ZR. Clinical, pathological and unusual MRI features of five synovial sarcomas in head and neck. Br J Radiol. 2015;88:20140843. doi: 10.1259/bjr.20140843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kambo JS, Richardson B, Ionescu DN, Tucker T, Kraushaar G. Primary pulmonary synovial sarcoma: a case report with unique and impressive computed tomography findings. Can Respir J. 2015;22:e1–e3. doi: 10.1155/2015/231043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Romeo S, Rossi S, Acosta Marín M, Canal F, Sbaraglia M, Laurino L, Mazzoleni G, Montesco MC, Valori L, Campo Dell’Orto M, et al. Primary Synovial Sarcoma (SS) of the digestive system: a molecular and clinicopathological study of fifteen cases. Clin Sarcoma Res. 2015;5:7. doi: 10.1186/s13569-015-0021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thway K, Fisher C. Synovial sarcoma: defining features and diagnostic evolution. Ann Diagn Pathol. 2014;18:369–380. doi: 10.1016/j.anndiagpath.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 68.Barham W, Frump AL, Sherrill TP, Garcia CB, Saito-Diaz K, VanSaun MN, Fingleton B, Gleaves L, Orton D, Capecchi MR, et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013;3:1286–1301. doi: 10.1158/2159-8290.CD-13-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trautmann M, Sievers E, Aretz S, Kindler D, Michels S, Friedrichs N, Renner M, Kirfel J, Steiner S, Huss S, et al. SS18-SSX fusion protein-induced Wnt/β-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene. 2014;33:5006–5016. doi: 10.1038/onc.2013.443. [DOI] [PubMed] [Google Scholar]
- 70.Brecht IB, Ferrari A, Int-Veen C, Schuck A, Mattke AC, Casanova M, Bisogno G, Carli M, Koscielniak E, Treuner J. Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer. 2006;46:11–17. doi: 10.1002/pbc.20502. [DOI] [PubMed] [Google Scholar]
- 71.Ferrari A, De Salvo GL, Brennan B, van Noesel MM, De Paoli A, Casanova M, Francotte N, Kelsey A, Alaggio R, Oberlin O, et al. Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005) Ann Oncol. 2015;26:567–572. doi: 10.1093/annonc/mdu562. [DOI] [PubMed] [Google Scholar]
- 72.Mnif H, Zrig M, Maazoun K, Sahnoun L, Bannour S, Koubaa M, Nouri A, Abid A. Congenital infantile fibrosarcoma of the forearm. Chir Main. 2011;30:148–151. doi: 10.1016/j.main.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 73.Russell H, Hicks MJ, Bertuch AA, Chintagumpala M. Infantile fibrosarcoma: clinical and histologic responses to cytotoxic chemotherapy. Pediatr Blood Cancer. 2009;53:23–27. doi: 10.1002/pbc.21981. [DOI] [PubMed] [Google Scholar]
- 74.Akyüz C, Küpeli S, Varan A, Gedikoglu G, Yalçin B, Kutluk T, Büyükpamukçu M. Infantile fibrosarcoma: retrospective analysis of eleven patients. Tumori. 2011;97:166–169. doi: 10.1177/030089161109700206. [DOI] [PubMed] [Google Scholar]
- 75.Orbach D, Rey A, Cecchetto G, Oberlin O, Casanova M, Thebaud E, Scopinaro M, Bisogno G, Carli M, Ferrari A. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol. 2010;28:318–323. doi: 10.1200/JCO.2009.21.9972. [DOI] [PubMed] [Google Scholar]
- 76.Sulkowski JP, Raval MV, Browne M. Margin status and multimodal therapy in infantile fibrosarcoma. Pediatr Surg Int. 2013;29:771–776. doi: 10.1007/s00383-013-3318-4. [DOI] [PubMed] [Google Scholar]
- 77.Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol. 2002;26:823–835. doi: 10.1097/00000478-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 78.Finke NM, Lae ME, Lloyd RV, Gehani SK, Nascimento AG. Sinonasal desmoplastic small round cell tumor: a case report. Am J Surg Pathol. 2002;26:799–803. doi: 10.1097/00000478-200206000-00016. [DOI] [PubMed] [Google Scholar]
- 79.Jian Z, Shaohong H, Wenzhao Z, Lijia G. Misdiagnosed desmoplastic small round cell tumor of the pleura: case report and literature review. J Formos Med Assoc. 2014;113:60–61. doi: 10.1016/j.jfma.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 80.Rao P, Tamboli P, Fillman EP, Meis JM. Primary intra-renal desmoplastic small round cell tumor: expanding the histologic spectrum, with special emphasis on the differential diagnostic considerations. Pathol Res Pract. 2014;210:1130–1133. doi: 10.1016/j.prp.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 81.Yue X, Wang JZ, Tian Y, Wang KJ. Paratesticular desmoplastic small round cell tumor with metastasis: a report of two cases. Kaohsiung J Med Sci. 2014;30:104–105. doi: 10.1016/j.kjms.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 82.Gerald WL, Rosai J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol. 1989;9:177–183. doi: 10.3109/15513818909022347. [DOI] [PubMed] [Google Scholar]
- 83.Lee YS, Hsiao CH. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical and molecular study of four patients. J Formos Med Assoc. 2007;106:854–860. doi: 10.1016/S0929-6646(08)60051-0. [DOI] [PubMed] [Google Scholar]
- 84.Ordóñez NG, el-Naggar AK, Ro JY, Silva EG, Mackay B. Intra-abdominal desmoplastic small cell tumor: a light microscopic, immunocytochemical, ultrastructural, and flow cytometric study. Hum Pathol. 1993;24:850–865. doi: 10.1016/0046-8177(93)90135-4. [DOI] [PubMed] [Google Scholar]
- 85.Lal DR, Su WT, Wolden SL, Loh KC, Modak S, La Quaglia MP. Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg. 2005;40:251–255. doi: 10.1016/j.jpedsurg.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 86.Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and outcomes of desmoplastic small round cell tumor: results from the surveillance, epidemiology, and end results database. J Cancer Epidemiol. 2014;2014:680126. doi: 10.1155/2014/680126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saab R, Khoury JD, Krasin M, Davidoff AM, Navid F. Desmoplastic small round cell tumor in childhood: the St. Jude Children’s Research Hospital experience. Pediatr Blood Cancer. 2007;49:274–279. doi: 10.1002/pbc.20893. [DOI] [PubMed] [Google Scholar]
- 88.Frezza AM, Benson C, Judson IR, Litiere S, Marreaud S, Sleijfer S, Blay JY, Dewji R, Fisher C, van der Graaf W, et al. Pazopanib in advanced desmoplastic small round cell tumours: a multi-institutional experience. Clin Sarcoma Res. 2014;4:7. doi: 10.1186/2045-3329-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Araújo RA, Araújo BJ. Desmoplastic small round cell tumor: report of 2 cases treated with chemotherapy alone or in combination with bevacizumab. Case Rep Oncol. 2014;7:102–108. doi: 10.1159/000359997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fan HS, I’Ons B, McConnell R, Kumar V, Alzahrani S, Morris DL. Peritonectomy and hyperthermic intraperitoneal chemotherapy as treatment for desmoplastic small round cell tumour. Int J Surg Case Rep. 2015;7C:85–88. doi: 10.1016/j.ijscr.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhagat P, Bal A, Das A, Singh N, Singh H. Pulmonary inflammatory myofibroblastic tumor and IgG4-related inflammatory pseudotumor: a diagnostic dilemma. Virchows Arch. 2013;463:743–747. doi: 10.1007/s00428-013-1493-2. [DOI] [PubMed] [Google Scholar]
- 92.Nagarajan S, Jayabose S, McBride W, Prasadh I, Tanjavur V, Marvin MR, Rodriguez-Davalos MI. Inflammatory myofibroblastic tumor of the liver in children. J Pediatr Gastroenterol Nutr. 2013;57:277–280. doi: 10.1097/MPG.0b013e31829e0b3b. [DOI] [PubMed] [Google Scholar]
- 93.Pradhan MR, Ranjan P, Rao RN, Chipde SS, Pradhan K, Kapoor R. Inflammatory myofibroblastic tumor of the urinary bladder managed by laparoscopic partial cystectomy. Korean J Urol. 2013;54:797–800. doi: 10.4111/kju.2013.54.11.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Groenveld RL, Raber MH, Oosterhof-Berktas R, Eijken E, Klaase JM. Abdominal inflammatory myofibroblastic tumor. Case Rep Gastroenterol. 2014;8:67–71. doi: 10.1159/000360843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ernst CW, Van Der Werff Ten Bosch J, Desprechins B, de Mey J, De Maeseneer M. Malignant transformation of an abdominal inflammatory myofibroblastic tumor with distant metastases in a child. JBR-BTR. 2011;94:78–80. doi: 10.5334/jbr-btr.500. [DOI] [PubMed] [Google Scholar]
- 96.Ak G, Jaaback K, Yin H, Maley P, Scurry J. Malignant inflammatory myofibroblastic tumour of the uterus. Pathology. 2015;47:380–381. doi: 10.1097/PAT.0000000000000260. [DOI] [PubMed] [Google Scholar]
- 97.Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, Pinkus GS, Xiao S, Yi ES, Fletcher CD, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157:377–384. doi: 10.1016/S0002-9440(10)64550-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bridge JA, Kanamori M, Ma Z, Pickering D, Hill DA, Lydiatt W, Lui MY, Colleoni GW, Antonescu CR, Ladanyi M, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001;159:411–415. doi: 10.1016/S0002-9440(10)61711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cools J, Wlodarska I, Somers R, Mentens N, Pedeutour F, Maes B, De Wolf-Peeters C, Pauwels P, Hagemeijer A, Marynen P. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2002;34:354–362. doi: 10.1002/gcc.10033. [DOI] [PubMed] [Google Scholar]
- 100.Debiec-Rychter M, Marynen P, Hagemeijer A, Pauwels P. ALK-ATIC fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;38:187–190. doi: 10.1002/gcc.10267. [DOI] [PubMed] [Google Scholar]
- 101.Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M, Zuppan C, Bridge JA. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;37:98–105. doi: 10.1002/gcc.10177. [DOI] [PubMed] [Google Scholar]
- 102.Panagopoulos I, Nilsson T, Domanski HA, Isaksson M, Lindblom P, Mertens F, Mandahl N. Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer. 2006;118:1181–1186. doi: 10.1002/ijc.21490. [DOI] [PubMed] [Google Scholar]
- 103.Takeuchi K, Soda M, Togashi Y, Sugawara E, Hatano S, Asaka R, Okumura S, Nakagawa K, Mano H, Ishikawa Y. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res. 2011;17:3341–3348. doi: 10.1158/1078-0432.CCR-11-0063. [DOI] [PubMed] [Google Scholar]
- 104.Karnak I, Senocak ME, Ciftci AO, Cağlar M, Bingöl-Koloğlu M, Tanyel FC, Büyükpamukçu N. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg. 2001;36:908–912. doi: 10.1053/jpsu.2001.23970. [DOI] [PubMed] [Google Scholar]