

Abstract
Background
Activation of systemic innate immunity is critical in the chain of events leading to restenosis. LABR-312 is a novel compound that transiently modulates circulating monocytes, reducing accumulation of these cells at vascular injury sites and around stent struts. The purpose of the study was to examine the safety and efficacy of a single intravenous bolus of LABR-312 in reducing restenosis in patients treated for coronary narrowing. Patient response was examined in light of differential inflammatory states as evidenced by baseline circulating monocyte levels, diabetes mellitus, and acute coronary syndrome.
Methods
BLAST is a Phase II prospective, randomized, multicenter, double-blind, placebo-controlled trial that assessed the safety and efficacy of LABR-312. Patients were randomized to receive LABR-312 at 2 dose levels or placebo as an intravenous infusion during percutaneous coronary intervention and bare metal stent implantation. The primary end point was mean angiographic in-stent late loss at 6 months.
Results
Patients (N = 225) were enrolled at 12 centers. There were no safety concerns associated with the study drug. For the overall cohort, there were no differences between the groups in the primary efficacy end point (in-stent late loss of 0.86 ± 0.60 mm, 0.83 ± 0.57 mm, and 0.81 ± 0.68 mm for the placebo, low-dose, and high-dose group, respectively; P = not significant for all comparisons). In the prespecified subgroups of patients with a baseline proinflammatory state, patients with diabetes mellitus, and patients with high baseline monocyte count, there was a significant treatment effect.
Conclusions
Intravenous administration of LABR-312 to patients undergoing percutaneous coronary intervention is safe and effectively modulates monocyte behavior. The average late loss did not differ between the treatment and placebo groups. However, in the inflammatory patient group with baseline monocyte count higher than the median value, there was a significant reduction in late loss with LABR-312.
The intimal hyperplastic reactions to endovascular implants and their clinical consequence, restenosis, have been appreciated for years; yet critical questions still remain as to why they occur, whether they can be controlled, and why certain subpopulations of patients suffer most. Of the most important clinical questions are those that center about understanding whether restenosis is the same problem for all or a unique process in high-risk patients. This issue has profound ramifications. Inherent in considering this issue is understanding if there is one driving mechanism, one common cause, and therefore potentially one universal target to combat the disease. Alternatively, restenosis, like many clinical syndromes, may represent a spectrum of responses to injury that involve a number of pathobiologic events that dominate to different degrees in different patients.1 If this is the case, then specific therapies might be designed for specific patients; and no one therapy should suffice for every person.
We now report on a clinical trial that examines whether a biologic identifier can delineate patient populations at risk, and response to therapy.
Much like native atherosclerosis, restenosis is deeply rooted within inflammation.2-6 Experimental and human studies indicate overactivation of circulating monocytes after vascular injury, which contributes to neointimal hyperplasia.7,8 LABR-312 (BIOrest, Northern Industrial Park, Yavneh, Israel) is a unique intravenous formulation designed to reduce restenosis (Figure 1) via site-specific anti-inflammatory and antiproliferative activity. The mechanism of action of LABR 312 has been studied extensively and described elsewhere.9-11 Briefly, transient modulation of circulating monocytes for several days follows a single systemic injection (Figure 2, A), permanently reducing monocyte accumulation at vascular injury sites and around stent struts, attenuating downstream signaling, and sustaining a decreased neoin-timal hyperplasia.9-15 The unique monocyte-targeting mechanism of action completely spares endothelial cells, allowing normal healing and standard dual-anti-platelet therapy (Figure 2, B).
Figure 1.
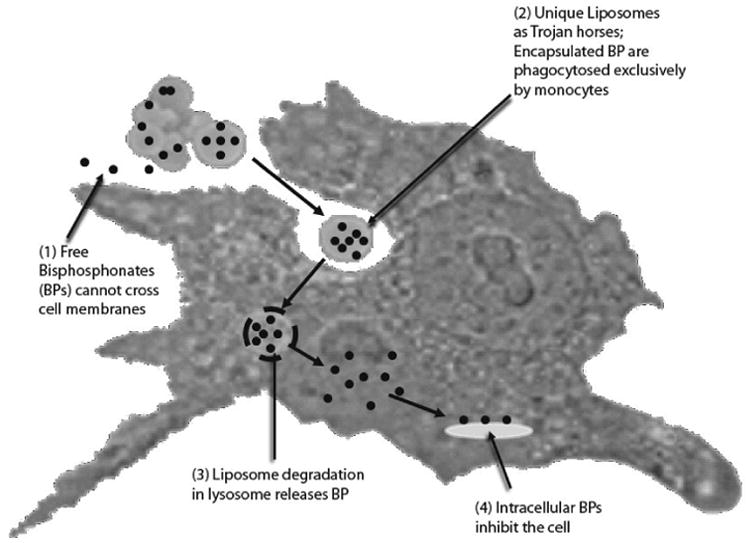
LABR-312 mechanism of action. Encapsulation in unique liposomes achieves targeting to phagocytic cells.
Figure 2.
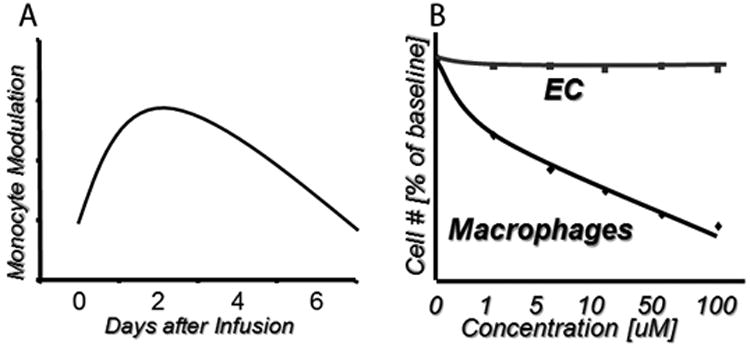
LABR-312 properties. A, Modulation of monocytic behavior (eg, number, activation status, cytokine expression) after a single injection peaks at 2 to 3 days and lasts for up to 1 week. B, Targeting via phagocytosis allows specificity to monocytes/macrophages while sparing endothelial cells.
The BLAST trial examined the safety and efficacy of a single intravenous bolus of LABR-312 in modulating monocyte function in a controlled and transient fashion. Subsequent angiographic late lumen loss at 6 months was assessed in patients treated for de novo stenotic native coronary atherosclerotic lesions with bare metal stent (BMS) implantation. Patient response was also examined in light of differential inflammatory states as evidenced by circulating monocyte levels, diabetes mellitus (DM), and acute coronary syndromes (ACS).
The aim was to examine if dichotomization on the basis of mechanism of action rather than patient demographics or lesion characteristics would correlate with treatment effect and offer a specific means of codifying patients and tailoring therapy for personalized medicine. Specifically, we examined if inflammation could be used as a unique restenosis risk identifier, clinical differentiator, and marker of response to disease modulation.
Methods
Study design and population
BLAST is a Phase II dose-finding, randomized, placebo-controlled, multicenter, prospective, double-blind clinical study conducted at 12 medical centers in Israel. The study protocol was approved by the ethics committee at each participating center and was conducted according to the principles of the Declaration of Helsinki. All patients provided written, informed consent for participation in this trial.
Participants enrolled had symptomatic ischemic heart disease from de novo coronary artery lesions with stable or unstable angina pectoris and baseline cardiac troponin of up to 3 times the upper limit of normal. Two or fewer lesions in 1 or 2 coronary arteries were treated with reference vessel diameter(s) 2.5 to 3.5 mm and lesion length(s) ≤30 mm. Standard inclusion/ exclusion criteria were used (see protocol). Clinically complex patients were specifically recruited to enrich the trial population with DM and unstable coronary syndromes. Patients were randomized 1:1:1 to receive low-dose LABR-312 (0.001 mg), high-dose LABR-312 (0.01 mg), or placebo (isotonic sodium chloride solution) (Figure 3).
Figure 3.
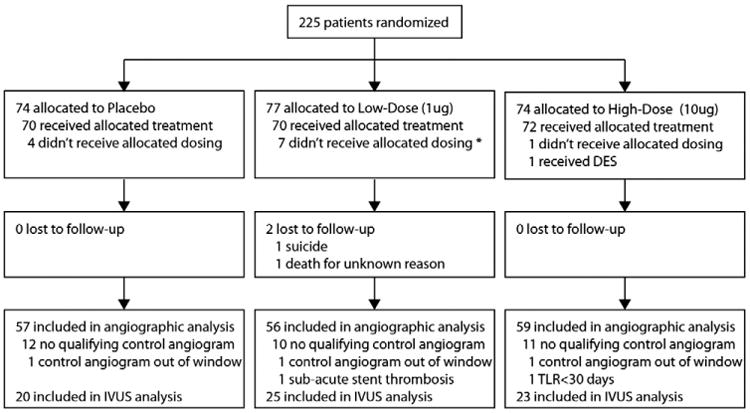
Trial profile. No reliable data are available regarding screening.
Active drug and placebo were supplied to the participating centers in identical, unmarked (except for serial number) vials. The concentration of the low dose was adjusted such that each vial contained identical volume. The absence of any other identifying features ensured that the centers were completely blinded as to which vial contained which treatment (or placebo). Neither the clinical sites nor the sponsor of the study held the master table that identified the contents of each vial. The master table was held at the central data management facility. Vials were supplied to the sites in groups of 6. Each group contained 2 high-dose, 2 low-dose, and 2 placebo vials. When a patient was recruited, the site would arbitrarily choose 1 of the 6 vials to open, thus ensuring randomization and balancing on a per-site basis. When all 6 vials had been used, another packet of 6 would be supplied to the site.
Stenting procedure and drug administration
To reduce variability, all patients received the Presillion baremetal stent (Medinol, Tel Aviv, Israel). Percutaneous coronary intervention (PCI), stenting, and adjunctive pharmacotherapy were performed according to individual site standard of care. Balloon predilatation was mandatory. Vascular closure devices were not allowed. Protocol-mandated dual-antiplatelet therapy was for at least 1 month postprocedure.
Randomization occurred during the procedure (after verification of angiographic inclusion/exclusion criteria and before any intervention). Drug was administered intravenously over 2 hours through a peripheral catheter within 30 minutes of predilatation.
Patient follow-up and data management
Patients were monitored in-hospital for at least 24 hours postprocedure. Clinical follow-up was at 30 days, 6 months, and yearly for 5 years (ongoing). Angiographic follow-up was mandated for all patients at 6 months. The first 110 patients (representing 50% of the cohort) recruited at sites where intravascular ultrasonography (IVUS) was available also underwent IVUS at follow-up postprocedure and at 6 months. All bloods for the monocyte-level analyses were analyzed centrally by a Fluorescence-activated cell sorting core laboratory.
There was 100% clinical monitoring by an independent monitoring group. All serious adverse events and all events considered by the investigators as at least possibly related to the study drug were adjudicated by a blinded, independent clinical events committee. An independent data and safety monitoring board met frequently and had access to all study data (blinded or unblinded upon request). All data were sent for analysis to independent consulting biostatisticians. Independent core laboratories analyzed all angiograms, IVUSs, and electrocardiograms.
Study end points
The primary efficacy end point was mean in-stent late loss, defined as the difference in minimal lumen diameter (MLD) between postprocedure and 6-month follow-up, measured by quantitative coronary angiography, on a per-patient basis for the per-protocol patient set. Secondary end points included major adverse cardiac events (MACE), target lesion(s) revascularization (TRL), and various angiographic and IVUS measures (see protocol).
Prespecified subgroup analysis
Subgroup analysis investigated the consistency in treatment effect. All subgroups were prespecified in the study's Statistical Analysis Plan (SAP). Aside from accepted generalized demographics and lesion characteristics, patients were divided on the basis of mechanistic determinants of underlying pathophysiology or drug effect. Mechanistic delineation into subgroups included DM, ACS, and inflammatory state as determined by baseline monocyte count.
Statistical analysis
All statistical tests (SAS Version 8.2; SAS, Cary, NC) and CIs were performed at P = .05 (2-sided) except as otherwise specified. Treatment group comparability was evaluated with respect to all clinically relevant demographic and baseline disease characteristics. Continuous variables were analyzed by one-way analysis of variance. Categorical variables were tested using Pearson χ2 test for contingency tables. Unless otherwise stated, all values reported are mean ± SD.
For the primary end point analysis, the pairwise comparison of each of the dose groups versus placebo was made at the α = .05 (2-sided) significance level. No adjustments were performed for the multiple comparisons given the exploratory nature of this study. Two independent-sample t tests were used for the main comparison.
Subgroup analyses were carried out to investigate the consistency in treatment effects on the primary end point between preselected variables. Two-way analysis of variance with interaction was used to detect differences in treatment effects between subgroups. The model included treatment, subgroup variables, and “treatment-by-subgroup variable” interactions as factors. For each treatment group, post hoc goodness-of-fit tests of the late loss distribution to the normal distribution were carried out using the Anderson-Darling statistic.16
A fund from BIOrest Ltd was used to support this clinical trial.
The authors of this manuscript are solely responsible for the design and conduct of this study, all study analyses, the drafting and editing of the paper, and its final contents.
Results
Patients and enrollment, safety, angiographic follow-up data, and restenosis
Between September 2008 and December 2009, 225 patients were enrolled at 12 sites (see online Appendix A). There were no statistically significant differences in patient demographics and in lesion or procedural characteristics (Tables I and II) between the 3 groups (except as noted for age). Angiographic follow-up data were available for 59 (80%) patients treated with high-dose LABR-312, 56 (73%) in the low-dose LABR-312 group, and 57 (77%) in the placebo group (P = not significant [NS]). Median follow-up time for the primary end point was 182 days.
Table I. Baseline characteristics.
| Placebo (n = 57) | Low dose (n = 56) | High dose (n = 59) | |
|---|---|---|---|
| Age (years ± SD) | 58.1 ± 8.2 | 62.3 ± 10.2 | 60.1 ± 9.4 |
| Male | 50 (87.7%) | 51 (91.1%) | 51 (86.4%) |
| Previous MI | 17 (30.4%) | 11 (19.6%) | 14 (23.7%) |
| Previous coronary artery bypass graft surgery | 4 (7.0%) | 4 (7.1%) | 3 (5.1%) |
| DM | 22 (38.6%) | 19 (33.9%) | 17 (28.8%) |
| Hypertension | 38 (66.7%) | 43 (76.8%) | 41 (69.5%) |
| Hypercholesterolemia | 43 (75.4%) | 49 (89.1%) | 50 (86.2%) |
| Premature CAD in 1st-degree relative | 20 (41.7%) | 19 (37.3%) | 21 (45.7%) |
| Current smoker | 23 (42.6%) | 14 (25.5%) | 21 (36.2%) |
| Previous PCI | 22 (38.6%) | 22 (39.3%) | 18 (30.5%) |
| Peripheral vascular disease | 2 (3.5%) | 4 (7.1%) | 4 (6.8%) |
| Chronic renal failure | 1 (1.8%) | 5 (8.9%) | 1 (1.7%) |
| Unstable angina | 38 (66.7%) | 39 (69.6%) | 31 (53.4%) |
| LVEF % (mean ± SD) | 58.3% ±9.5% | 56.3% ±8.4% | 58.5% ±8.9% |
| Vessel location | |||
| RCA | 17 (29.8%) | 13 (23.2%) | 15 (25.4%) |
| LAD | 22 (38.6%) | 26 (46.4%) | 23 (39.0%) |
| LCX | 18 (31.6%) | 17 (30.4%) | 21 (35.6%) |
| LMCA | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Diffuse lesion length (≥20 mm) | 5 (8.8%) | 2 (3.6%) | 5 (8.5%) |
| Thrombus | 2 (3.5%) | 0 (0.0%) | 2 (3.4%) |
| Moderate to severe calcification | 8 (14.0%) | 12 (21.4%) | 14 (23.7%) |
| TIMI flow | |||
| 0 | 1 (1.8%) | 0 (0.0%) | 1 (1.7%) |
| 1 | 1 (1.8%) | 2 (3.6%) | 1 (1.7%) |
| 2 | 5 (8.8%) | 7 (12.5%) | 5 (8.5%) |
| 3 | 50 (87.7%) | 47 (83.9%) | 52 (88.1%) |
| Total occlusion | 2 (3.5%) | 2 (3.6%) | 2 (3.4%) |
Data are patients (percentage) except where noted. P = NS for all comparisons except for age (P = .016 low dose vs placebo.
MI, Myocardial infarction; CAD, coronary artery disease; LVEF, left ventricular ejection fraction; RCA, right coronary artery; LAD, left anterior descending artery; LCX, left circumflex artery; LMCA, left main coronary artery.
Table II. Angiographic and procedural characteristics.
| Placebo (n = 57) | Low dose (n = 56) | High dose (n = 59) | |
|---|---|---|---|
| Preprocedure | |||
| Reference vessel diameter (mm) | 2.75 ± 0.46 | 2.67 ± 0.47 | 2.84 ± 0.41 |
| MLD (mm) | 0.77 ± 0.28 | 0.76 ± 0.31 | 0.85 ± 0.33 |
| % Stenosis | 71.72 ± 9.52 | 71.92 ± 9.11 | 70.24 ± 9.54 |
| Lesion length (mm) | 12.14 ± 4.58 | 12.12 ± 4.62 | 13.14 ± 5.47 |
| No. of lesions per patient (mean ± SD) | 1.16 ± 0.37 | 1.11 ± 0.31 | 1.20 ± 0.45 |
| Postprocedure | |||
| In-stent MLD (mm) | 2.63 ± 0.48 | 2.59 ± 0.49 | 2.67 ± 0.38 |
| In-stent % stenosis | 6.23 ± 5.94 | 4.64 ± 9.82 | 6.49 ± 6.81 |
| Acute gain (mm) | 1.85 ± 0.45 | 1.83 ± 0.43 | 1.82 ± 0.35 |
Data are mean ± SD. P = NS for all comparisons.
There were no statistically significant differences in safety between either drug treatment group or the placebo, and no specific safety concerns associated with the study drug at either dose (Table III). Because of the recruitment emphasis on complex patients, the trial population was enriched for DM and ACS, accounting for the relatively high total MACE values in all groups. There were no effect on neutrophil or platelet count and no differences seen in acute, subacute, or chronic liver function test results.
Table III. Main safety results to 180 days.
| Placebo | Low dose | P | High dose | P | |
|---|---|---|---|---|---|
| MACE | 26.5% | 25.4% | .88 | 20.8% | .43 |
| Death | 0% | 2.8% | .16 | 0% - | - |
| MI* | 22.1% | 11.3% | .09 | 12.5% | .13 |
| Clinically driven TLR | 5.9% | 15.5% | .07 | 12.5% | .18 |
| SAE probably related to drug | 1.4% (1) | 2.7% (2) | 1.4% (1) |
Data are percentage of patients from safety analysis data set.
SAE, Serious adverse events.
MI excluding periprocedural = 1.8%, 1.8%, and 1.7% for placebo, low-dose, and high-dose group, respectively.
There were no statistically significant differences in primary efficacy between the 3 groups in any one of the commonly measured parameters based on average values (Table IV). In-stent late loss (the primary end point) was generally low compared with historical BMS values, yet not different between the treatment groups and placebo (0.86 ± 0.60 mm, 0.83 ± 0.57 mm, and 0.81 ± 0.68 mm for placebo, low-dose, and high-dose group, respectively; P = NS).
Table IV. Main efficacy results at 180 days.
| Placebo | Low dose | High dose | P | |
|---|---|---|---|---|
| In-stent LL (mm) | 0.86 ± 0.60 | 0.83 ± 0.57 | 0.81 ± 0.68 | NS |
| In-stent MLD (mm) | 177 ± 0.80 | 1.75 ± 0.81 | 1.87 ± 0.71 | NS |
| % DS | 36.64 ± 24.88 | 34.86 ± 27.16 | 33.29 ± 23.80 | NS |
| IVUS % volume obstruction | 24.2 ± 14.5 | 24.4 ± 11.6 | 26.3 ± 15.5 | NS |
Data are mean ± SD.
LL, Late loss.
The distribution curves for the primary end point differed significantly. The distribution for the placebo group is symmetric and follows the expectation for a normal (Gaussian) distribution (P > .2). In contrast, both treatment groups exhibit nonnormal distributions that are skewed to the left with a rightward tail, and peaks that fall well to the left of the comparator placebo peak. Both treatment distributions fail the mathematical test for normality (P < .005).
Analysis of the primary end point in prespecified subgroups
In the prespecified subgroup analysis, demographic parameters, lesion characteristics, and ACS did not distinguish treatment effect. For all of these variables, there were no statistically significant differences or any appreciable trends observed between the treatment and placebo groups on the primary end point. In contrast, DM patients showed a large numerical improvement (Table V); and inflammation was a specific marker of statistically significant effect with LABR-312 (Table V). In the placebo group, the DM patients had appreciably larger late loss values than nondiabetic persons as expected (1.05 ± 0.60 mm vs 0.74 ± 0.58 mm, DM vs non-DM, respectively). This 42% increase was erased in the low-dose (0.86 ± 0.56 mm vs 0.82 ± 0.58 mm) and in the high-dose groups (0.77 ± 0.62 mm vs 0.82 ± 0.71 mm, DM vs non-DM, respectively), resulting in a nearly 30% reduction in late loss in the DM group for high dose relative to placebo (P = .16).To investigate whether baseline inflammatory function had an effect on the response to LABR-312, the following pre hoc analysis was performed: The entire cohort was divided evenly into 2 subgroups based on their baseline monocyte count (median count = 539.5/μL). Patients whose monocyte count was higher than the median (n = 82) were termed the high-monocyte (presumably more inflammatory) group, whereas patients whose monocyte count was lower than the median (n = 83) were termed the low-monocyte (presumably less inflammatory) group. There were 32% and 37% diabetic patients in the low- and high-monocyte groups, respectively (34% overall in the study). No interaction was detected between diabetes status and monocyte count. In the high-monocyte patients, there was a statistically significant reduction in late loss of >30% in the high-dose group (Table V) relative to placebo.
Table V. Primary end point by baseline diabetes or monocyte count.
| In-stent late loss (mm) | Placebo | Low dose | P (low dose vs placebo) | High dose | P (high dose vs placebo) |
|---|---|---|---|---|---|
| By diabetes | |||||
| Nondiabetic participants | 0.74 ± 0.58 | 0.82 ± 0.58 | .57 | 0.82 ± 0.71 | .60 |
| Diabetic participants | 1.05 ± 0.60 | 0.86 ± 0.56 | .30 | 0.77 ± 0.62 | .16 |
| By baseline monocytes | |||||
| Low monocytes* | 0.67 ± 0.50 | 0.86 ± 0.57 | .24 | 0.86 ± 0.70 | .29 |
| High monocytes† | 1.00 ± 0.62 | 0.78 ± 0.60 | .18 | 0.67 ± 0.50 | .03 |
Data are mean ± SD.
Monocyte count less than the median preprocedure.
Monocyte count higher than the median preprocedure.
Discussion
From the first recognition of the long-term effects of percutaneous vascular interventions, the question has been raised as to how to consider patients with clinical restenosis.17-19 It was then and remains even now not clear as to whether all patients suffer from restenosis or if some patients are at greater risk and others relatively immune. We do not know if all patients are sensitive to therapy or if some are more sensitive and others more resistant—whether there is one drug for all or specific drugs for specific patients. The BLAST trial investigated some of these questions. In particular, we sought to determine if transient modulation of a specific cellular target, inflammatory monocytes, could elicit a permanent effect on vascular response to injury.
Why should inflammation dichotomize patients more effectively than other pre hoc differentiators? Unlike clinical risk factors, inflammation is a biological event with mechanistic undertones and is central to all processes that follow. Inflammation, and in particular the monocytic component, is directly correlative of vascular injury in controlled animal models. Monocyte infiltration directly drives thrombosis, vascular smooth muscle cell proliferation, and matrix remodeling. And yet, past attempts at modulating inflammation have not successfully limited restenosis. Local delivery of anti-inflammatory agents has had minimal effects, as inflammation, even when locally active, is replenished from systemic circulating cells. Systemic infusions often lack local specificity. Most notably, the IMPRESS study, which used systemically administered corticosteroids, only showed a modest effect in a small, highly proinflammatory subpopulation.17,18
In contrast to the nonspecific effects of corticosteroids, LABR-312 has the potential to selectively alter the local inflammatory reaction to endovascular stent implants through systemic targeting of monocytes/ macrophages.11-15, 19, 20 Systemic inhibition of mono-cytes with LABR-312 was sought to prevent accumulation of these cells at sites of injury like stent struts and resolve after a week. The specificity to monocytes preserves endothelial cells and normal healing, minimizing adverse effects.
For all these reasons, we hypothesized that systemic delivery of LABR-312 in patients undergoing BMS implantation will attenuate restenosis.
We show that the clinical efficacy of systemic LABR-312 was observed only in patient population segments but not the entire cohort studied. On average, the late lumen loss values did not differ between the treatment groups and the placebo group. The response to modulation of inflammation was effective only in those patients deemed inflammatory and identified as such pre hoc. In the inflammatory patient group, there was a statistically significant and clinically meaningful reduction in late loss with LABR-312.
The late loss distributions might be more informative than the average values. These distribution curves clearly show important differences between the 3 dose groups. That the distribution curve for the placebo group was normal (Gaussian), whereas the ones for both treatment groups were clearly not, provides support for the notion of a treatment effect of the drug, despite the mean values being nearly identical. The asymmetric appearance of the treatment-group distributions would seem to indicate that there was a differential response to this therapy among patients. Although some evidence exists to support an outright bimodal distribution in the treatment groups, it is impossible to conclude based on the current data set whether patients could be separated into “responders” and “nonresponders” or whether there was a continuous spectrum of effect.
The segmentation of patients on the basis of DM is intriguing in 2 respects—the DM patients in the treatment group did considerably better than the DM in the placebo group, and treated DM patients did no worse than treated non-DM patients. Despite the numerical differences, these effects did not reach statistical significance, likely as this study was not powered for the number of diabetic patients. Future studies may very well investigate this specific question. Statistical significance was achieved in the so-called high-monocytes or inflammatory group that included 50% of the patients per definition. The importance of this finding is amplified by the fact that, even in the high-monocytes subgroup, monocyte levels were within normal limits as set by the individual hospital laboratories. And yet, a large (30%), clinically meaningful, and statistically significant effect of the drug was shown in this group, indicating that this anti-inflammatory drug indeed works best in proinflammatory patients.21-23
Conclusions
Systemic administration of LABR-312 to patients undergoing PCI is safe and effectively modulates monocyte behavior. However, it did not reduce the average late loss compared with placebo in the entire study cohort. In the inflammatory patient subgroup, there was a significant reduction in late loss with LABR-312. We have shown that populations that are homogeneous in clinical presentation may not be homogeneous in treatment response and that mechanistic rather than clinical characteristics might be important determinant of response to therapy.
In the BLAST trial, inflammation was best at identifying patients at risk for disease and sensitivity to therapy. Proof that one could specify which patients are most at risk for disease and most amenable to a specific therapy may provide the potential for personalized medicine—selecting specific drugs for specific patients. Future clinical trials should focus on these issues in an attempt to more precisely describe just who might benefit from this type of therapy.
Acknowledgments
Disclosures: Funding: The trial sponsor, BIOrest Ltd, Israel, provided all funding for the study.
Appendix A. Participating medical centers and names of principal investigators for each center
Tel Aviv Medical Center, Tel Aviv, Israel: Shmuel Banai
Shaare Zedek Medical Center, Jerusalem, Israel: Yarron Almagor
Baruch Padeh Hospital, Poriya, Israel: Jonathan Hasin
Bnei Zion Hospital, Haifa, Israel: Uri Rosenschein
Rabin Medical Center, Petach Tiqua, Israel: Ran Kornowski
Sheba Medical Center, Ramat Gan, Israel: Victor Guetta
Lady Davis Carmel Hospital, Haifa, Israel: Bazil Lewis
Meir Hospital, Kfar Saba, Israel: Moris Mosseri
Kaplan Hospital, rehovot, Israel: Oded Ayzenberg
Hillel Yaffe Hospital, Hadera, Israel: Aharon Frimerman
Western Galilee Hospital, Nahariya, Israel: Shaul Atar
Study management: Medinol Ltd.
Data management, clinical event committee, and data safety monitoring board coordination: Harvard Clinical research Institute, Boston, MA, USA
Angiographic core laboratory: Cardiovascular Research Foundation, New York, NY, USA
IVUS core laboratory: Stanford University Medical Center, Stanford, CA, USA
Arrhythmia and ECG core laboratory: Harvard Clinical Research Institute, Boston, MA, USA
FACS core laboratory: BIOrest Ltd, Yavneh, Israel
Study Monitoring: GCP Clinical Monitoring Ltd, Rosh Ha'Ayn, Israel.
Appendix B.
For the Biorest Liposomal Alendronate with Stenting sTudy - BLAST, see http://clinicaltrials.gov/ct2/show/NCT00739466?term=BLAST+study&rank=10.
Footnotes
Clinical Trial Registration—URL: http://www.ClinicalTrials.gov.Unique identifiers: NCT00739466.
References
- 1.Lehmann KG, Melkert R, Serruys PW. Contributions of frequency distribution analysis to the understanding of coronary restenosis, a reappraisal of the Gaussian curve. Circulation. 1996;93:1123–32. doi: 10.1161/01.cir.93.6.1123. [DOI] [PubMed] [Google Scholar]
- 2.Fuster JJ, Fernández P, González-Navarro H, et al. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res. 2010;86(2):254–64. doi: 10.1093/cvr/cvp363. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK. Atherosclerosis—an immune disease: the Anitschkov lecture 2007. Atherosclerosis. 2009;202(1):2–10. doi: 10.1016/j.atherosclerosis.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Rogers C, Welt FG, Karnovsky MJ, et al. Monocyte recruitment and neointimal hyperplasia in rabbits. Coupled inhibitory effects of heparin. Arterioscler Thromb Vasc Biol. 1996;16(10):1312–8. doi: 10.1161/01.atv.16.10.1312. [DOI] [PubMed] [Google Scholar]
- 5.Edelman ER, Rogers C. Pathobiologic responses to stenting. Am J Cardiol. 1998;81(7A):4E–6E. doi: 10.1016/s0002-9149(98)00189-1. [DOI] [PubMed] [Google Scholar]
- 6.Costa MA, Simon DI. Molecular basis of restenosis and drug-eluting stents. Circulation. 2005;111(17):2257–73. doi: 10.1161/01.CIR.0000163587.36485.A7. [DOI] [PubMed] [Google Scholar]
- 7.Schober A, Weber C. Mechanisms of monocyte recruitment in vascular repair after injury. Antioxid Redox Signal. 2005;7(9-10):1249–57. doi: 10.1089/ars.2005.7.1249. [DOI] [PubMed] [Google Scholar]
- 8.Toutouzas K, Colombo A, Stefanadis C. Inflammation and restenosis after percutaneous coronary interventions. Eur Heart J. 2004;25(19):1679–87. doi: 10.1016/j.ehj.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Danenberg HD, Fishbein I, Gao J, et al. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- 10.Huitinga I, Damoiseaux JG, van Rooijen N, et al. Liposome mediated affection of monocytes. Immunobiology. 1992;185(1):11–9. doi: 10.1016/S0171-2985(11)80313-X. [DOI] [PubMed] [Google Scholar]
- 11.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174(1-2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.Danenberg HD, Golomb G, Groothuis A, et al. Liposomal alendronate inhibits systemic innate immunity and reduces in-stent neointimal hyperplasia in rabbits. Circulation. 2003;108:2798–804. doi: 10.1161/01.CIR.0000097002.69209.CD. [DOI] [PubMed] [Google Scholar]
- 13.Cohen-Sela E, Dangoor D, Epstein H, et al. Nanospheres of a bisphosphonate attenuate intimal hyperplasia. J Nanosci Nanotechnol. 2006;6(9-10):3226–34. doi: 10.1166/jnn.2006.428. [DOI] [PubMed] [Google Scholar]
- 14.Cohen-Sela E, Rosenzweig O, Gao J, et al. Alendronate-loaded nanoparticles deplete monocytes and attenuate restenosis. J Control Release. 2006;113(1):23–30. doi: 10.1016/j.jconrel.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Danenberg HD, Fishbein I, Epstein H, et al. Systemic depletion of macrophages by liposomal bisphosphonates reduces neointimal formation following balloon-injury in the rat carotid artery. J Cardiovasc Pharmacol. 2003;42(5):671–9. doi: 10.1097/00005344-200311000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Stephens MA. EDF statistics for goodness of fit and some comparisons. J Am Stat Assoc. 1974;69:730–7. [Google Scholar]
- 17.Weintraub WS. The pathophysiology and burden of restenosis. Am J Cardiol. 2007;100(5A):3K–9K. doi: 10.1016/j.amjcard.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Melikian N, Wijns W. Drug-eluting stents: a critique. Heart. 2008;94(2):145–52. doi: 10.1136/hrt.2005.066993. [DOI] [PubMed] [Google Scholar]
- 19.Douglas JS. Pharmacologic approaches to restenosis prevention. Am J Cardiol. 2007;100(5A):10K–6K. doi: 10.1016/j.amjcard.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Ferrero V, Tomai F, Versaci F, et al. Long-term results of immunosuppressive oral prednisone after coronary angioplasty in non-diabetic patients with elevated C-reactive protein levels. Euro-Intervention. 2009;5(2):250–4. doi: 10.4244/eijv5i2a39. [DOI] [PubMed] [Google Scholar]
- 21.Versaci F, Gaspardone A, Tomai F, et al. Immunosuppressive Therapy for the Prevention of Restenosis after Coronary Artery Stent Implantation (IMPRESS Study) J Am Coll Cardiol. 2002;40(11):1935–42. doi: 10.1016/s0735-1097(02)02562-7. [DOI] [PubMed] [Google Scholar]
- 22.Fleisch H. Bisphosphonates: mechanisms of action. Endocr Rev. 1998;19(1):80–100. doi: 10.1210/edrv.19.1.0325. [DOI] [PubMed] [Google Scholar]
- 23.Rodan GA. Mechanisms of action of bisphosphonates. Annu Rev Pharmacol Toxicol. 1998;38:375–88. doi: 10.1146/annurev.pharmtox.38.1.375. [DOI] [PubMed] [Google Scholar]