

Abstract
The DEAD-box-protein DDX5 is an ATP-dependent RNA helicase that is frequently overexpressed in various cancers and acts as a transcriptional co-activator of several transcription factors, including β-catenin. DDX5 is reported to be involved in cancer progression by promoting cell proliferation and epithelial–mesenchymal transition. However, the clinical significance and biological role of DDX5 in non-small-cell lung cancer (NSCLC) remain largely unknown. In this study, we examined the expression of DDX5 in clinical NSCLC samples, investigated its role in regulating NSCLC cell proliferation and tumorigenesis, and explored the possible molecular mechanism. We found that DDX5 was significantly overexpressed in NSCLC tissues as compared with the matched normal adjacent tissues. In addition, overexpression of DDX5 was associated with advanced clinical stage, higher Ki67 index, and shorter overall survival in NSCLC patients. Upregulation of DDX5 promoted proliferation of NSCLC cells in vitro and growth of NSCLC xenografts in vivo, whereas downregulation of DDX5 showed the opposite effects. Furthermore, DDX5 directly interacted with β-catenin, promoted its nuclear translocation, and co-activated the expression of cyclin D1 and c-Myc. β-catenin silencing significantly abrogated DDX5-induced cyclin D1 and c-Myc expression and proliferation in NSCLC cells. Interestingly, DDX5 and cyclin D1 expression followed positive correlation in the same set of NSCLC samples. These findings indicated that DDX5 played an important role in the proliferation and tumorigenesis of NSCLC cells by activating the β-catenin signaling pathway. Therefore, DDX5 may serve as a novel prognostic marker and potential therapeutic target in the treatment of NSCLC.
Keywords: β-catenin, cyclin D1, DDX5, non-small-cell lung cancer, proliferation
Lung cancer is the most frequently diagnosed cancer and the leading cause of cancer death in the world.1 Approximately 80% of newly diagnosed lung cancers are non-small-cell lung cancer (NSCLC). Surgery is the initial treatment for early-stage NSCLC patients, but even after complete resection, a substantial proportion of patients suffer from recurrence. Over the past decade, significant progress has been made in the treatment of NSCLC, however, the 5-year overall survival (OS) rate remains at approximately 15%.2,3 Therefore, it is urgent to uncover the molecular mechanisms involved in NSCLC progression, which may help to provide new prognostic biomarkers and therapeutic targets for NSCLC therapy.
The DEAD-box RNA helicase DDX5 is an ATP-dependent RNA helicase characterized by the sequence Asp-Glu-Ala-Asp (D-E-A-D) in the Walker B motif required for ATP binding/hydrolysis and RNA binding/unwinding activities.4 DDX5 is ubiquitously expressed in all dividing cells of different vertebrates and participates in nearly all aspects of RNA metabolism, including RNA splicing, translation, ribosome biogenesis, and miRNA processing.5,6 DDX5 may also act in the regulation of gene transcription as transcriptional co-regulators with estrogen receptor-α, p53, androgen receptor, and β-catenin.7–11 Increasing evidence suggests that DDX5 is overexpressed in a variety of malignancies, including colorectal cancer, breast cancer, prostate cancer, head and neck squamous cell carcinoma, glioma, and leukemia.9,10,12–19 DDX5 is implicated in cancer development and progression by functioning in several key cellular activities of cancer cells, such as proliferation, migration, cytoskeletal reorganization, and epithelial–mesenchymal transition (EMT).12–16 For example, DDX5 is frequently amplified in breast cancer and is required for cell proliferation by controlling the transcription of DNA replication genes in breast cancer cells harboring this amplification.16 In colorectal cancer, DDX5 may promote cell proliferation and EMT by interacting with Wnt–β-catenin signaling.14
Despite extensive published reports elucidating the role of DDX5 in promoting cancer progression, little is known about its involvement in NSCLC. In the present study, we examined the expression of DDX5 in NSCLC tissues and uncovered its role in regulating NSCLC cell proliferation and tumorigenesis. We further investigated the molecular mechanism with respect to β-catenin/cyclin D1 signaling in the context of DDX5-mediated NSCLC cell proliferation.
Materials and Methods
Cell lines and culture
Human NSCLC cell lines H520 and A549 were purchased from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM-F12 (for A549) or RPMI-1640 medium (for H520) supplemented with 10% FBS (Gibco, Grand Island, NY, USA), 100 μg/mL streptomycin, and 100 U/mL penicillin at 37°C in a humidified incubator with 5% CO2.
Patient information and tissue samples
This study was approved by the Ethics Committee of the Fourth Military Medical University (Xi’an, China) and prior consent was obtained from participating patients. A total of 120 paraffin-embedded NSCLC specimens and the paired normal adjacent tissues were collected in Tangdu Hospital (Xi’an, China) from 2007 to 2010. Another 56 freshly collected paired NSCLC tissues were frozen and stored in liquid nitrogen for further use. Clinical and pathological classification and stage were determined according to the American Joint Committee on Cancer criteria. Overall survival was calculated from the date of diagnosis to death.
Retroviral infection and siRNA transfection
DDX5 downregulation was achieved using the retroviral expression vector GV118 (Genechemistry, Shanghai, China). The target sequence was GAACTGCTCGCAGTACCAA. DDX5 expression construct (pReciever-Lv105-DDX5) was purchased from Genecopoeia (Guangzhou, China). Viruses were generated and used to infect target cells. Stable cell lines expressing DDX5 and DDX5-RNAi were selected with 0.5 μg/mL puromycin for 2 weeks. Cells were also infected with corresponding vectors as negative control. Small interfering RNA targeting β-catenin (siBcat) and its negative control (siCon) were obtained from Genechemistry. Transfection of siRNAs was carried out using Lipofectamine 2000 and Opti-MEM Reduced-Serum medium (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions.
Western blot analysis
Proteins were extracted using the M-PER mammalian total and nuclear protein extraction reagents kits with protease and phosphatase inhibitors (Thermo Fisher Scientific, Rockford, IL, USA). The protein concentration of the supernatant was determined by BCA assay kit (Thermo). Western blot analysis was carried out using the following antibodies: rabbit anti-DDX5, rabbit anti-β-catenin, rabbit anti-cyclin D1, rabbit anti-cMyc, rabbit anti-lamin B, and mouse anti-β-actin (Abcam, Cambridge, UK). Blots were quantified with ImageJ (National Institutes of Health, Bethesda, MD, USA).
Immunostaining
Immunohistochemistry (IHC) was carried out as previously reported.17 The following antibodies were used: anti-DDX5, anti-cyclin D1, and anti-Ki67 (CST, Danvers, MA USA). Immunohistochemical staining was assessed by combining the percentage of positively stained cells (0–100%) and the intensity of staining (0, no staining; 1, week staining; 2, moderate staining; 3, strong staining). The total score (H score) ranges from 0 to 300. The cut-off values of 50 and 30 were used for DDX5 and Ki67, respectively, to divide the samples into low expression and high expression groups. Immunofluorescence was carried out as previously described.18 Slips were incubated with rabbit anti-β-catenin antibody, labeled with Texas Red-conjugated goat anti-rabbit secondary antibody (Abcam, Cambridge, MA, USA). Cells were observed and imaged under a fluorescence microscope.
Absorbance assay
Briefly, cells seeded in 96-well plates were cultured for 24, 48, and 72 h, and further incubated with 5 mg/mL MTT (Sigma, St. Louis, MO, USA) for 4 h. The absorbance was measured at 570 nm on a microplate reader (BioTek Instruments, Winooski, VT, USA).
Incorporation assay
The EdU incorporation assay was carried out as previously described,18 using a Cell-Light EdU Apollo 488 In Vitro Imaging Kit (Ribobio, Guangzhou, China). EdU-labeled cells were counted in 10 randomly selected fields under a fluorescent microscope.
Colony formation assay
Cells were seeded into 60-mm cell culture dishes (400 cells per dish) and cultured in 10 mL complete medium for 10 days. Afterwards, cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet. The number of colonies was counted.
Nude mice study
Animal experiments were approved by the Laboratory Animal Ethics Committee of the Fourth Military Medical University. Human NSCLC H520 cells (3 × 106 cells suspended in 100 μL PBS) stably infected with DDX5 or DDX5-RNAi and their corresponding control vectors were injected s.c. into 4-week-old male nude mice (Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China). Tumors were dissected 4 weeks thereafter and tumor volume and mass were determined. All tumors were fixed and embedded. Expression of DDX5, Ki-67, and cyclin D1 was examined by IHC.
Immunoprecipitation
Immunoprecipitation was carried out using a Pierce classic IP kit (Thermo) according to the manufacturer’s instructions. Briefly, total proteins were extracted and quantified. A total of 500 μg proteins in 400 μL supernatants were incubated with 4 μg anti-DDX5 or anti-β-catenin antibodies under rotation for 12 h at 4°C. The beads were washed, eluted in sample buffer, and boiled for 10 min at 100°C. The immune complexes were subjected to Western blot analysis as described above using the indicated primary antibodies. Cell extract incubated with equal amount of PBS was used as a negative control, and submitted directly to immunoprecipitation and Western blot (beads lane).
Real-time PCR
Total RNA was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) and reverse transcribed and amplified with a QuantiTect SYBR Green PCR Kit (Takara, Dalian, China) as described by the manufacturer. The following primers were used: cyclin D1, 5′-CTGGCCATGAACTACCTGGAC-3′ and 5′-GGTCACACTTGATCACTCTGG-3′; c-Myc, 5′-AGCGACTCTGAGGAGGAACAAG-3′ and 5′-CCTGCCTCTTTTCCACAGAAA-3′; and GAPDH, 5′-GACTCATGACCACAGTCCATGC-3′ and 5′-AGAGGCAGGGATGATGTTCTG-3′.
Dual luciferase reporter assay
The human cyclin D1 promoter luciferase reporter construct (pGL3-cyclin D1) was purchased from Addgene (Cambridge, MA, USA). TOPflash and FOPflash constructs were obtained from Millipore (Billerica, MA, USA). DDX5 or DDX5-RNAi infected cells were transiently co-transfected with the indicated constructs and pRL-TK Renilla luciferase plasmid (Promega, Madison, WI, USA) using Lipofectamine 2000. Luciferase activity was determined 48 h after transfection using the Dual Luciferase Reporter Assay System (Promega), as specified by the manufacturer.
Statistical analysis
Statistical analysis was carried out using spss 18.0 software (SPSS, Chicago, IL, USA). Comparison of DDX5 expression in the paired tissues was carried out using the non-parametric Wilcoxon rank sum analysis. The correlation between DDX5 expression and the clinicopathological characteristics was analyzed using the χ2-test. Survival curves were plotted using the Kaplan–Meier method and compared with a log–rank test. Multivariate survival analysis was done by the Cox proportional hazard model. Numerical data were presented as mean ± SEM and compared with a two-tailed Student’s t-test. The association between DDX5 expression and cyclin D1 expression was analyzed using Spearman’s rank correlation test. P < 0.05 was considered statistically significant.
Results
Expression of DDX5 was elevated in human NSCLC samples
The expression of DDX5 was examined in NSCLC tissues and the matched adjacent normal tissues (ANT). Western blot analysis of 56 pairs of fresh tissues revealed that expression of DDX5 was significantly increased in NSCLC tissues in comparison with the ANT (Fig.1). Similarly, IHC analysis of 120 paired paraffin-embedded tissues suggested that DDX5 was significantly upregulated in NSCLC tissues as compared with the matched ANT from the same patient (Fig.1).
Figure 1.
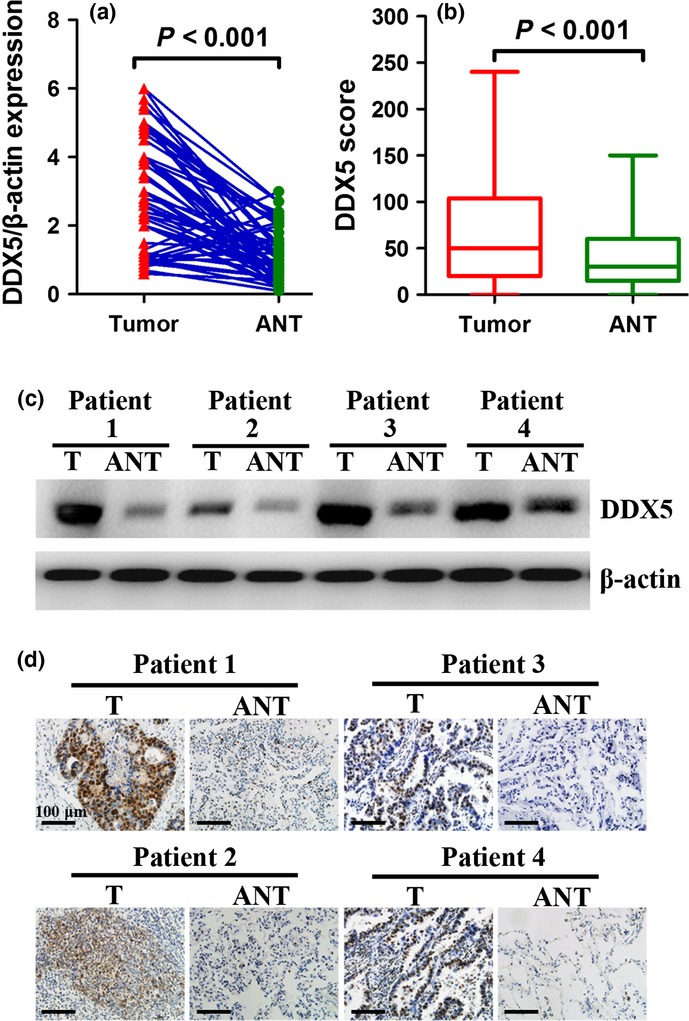
Expression of DDX5 is elevated in non-small-cell lung cancer (NSCLC) tissues. (a, c) Western blot analysis of DDX5 in 56 pairs of NSCLC tumors (T) and corresponding adjacent normal tissues (ANT). (b, d) Immunohistochemical staining of DDX5 in 120 cases of paraffin-embedded NSCLC tissues (T) and paired adjacent normal tissues (ANT). DDX5 is significantly overexpressed in NSCLC tissues in comparison with matched ANT.
Increased expression of DDX5 is correlated with clinicopathological features and poor prognosis of NSCLC patients
To investigate the clinical relevance of DDX5 expression, we analyzed the association between DDX5 expression and the clinicopathological features and prognosis of NSCLC patients. The 120 NSCLC patients were divided into high expression and low expression groups based on the immunostaining score of DDX5 with a cut-off value of 50. As shown in Table S1, expression of DDX5 was significantly associated with clinical stage (P < 0.001), tumor size (P < 0.001), and nodal status (P < 0.001). However, it was not associated with patient age, sex, smoking status, pathological grade, histologic type, or prior adjuvant treatment status. As DDX5 is reported to contribute to cancer cell proliferation, we further explored its relationship with the expression of Ki67. As shown in Figure2(a,b), expression of DDX5 was positively correlated with the Ki67 index in NSCLC (P < 0.001). Survival analysis revealed that patients with high expression of DDX5 had significantly shorter OS than those with low levels of DDX5 (P < 0.001; Fig.2c). The median OS in patients with low and high expression of DDX5 was 44 and 17 months, respectively. Moreover, univariate and multivariate analyses confirmed that clinical stage, nodal status, prior adjuvant therapy, and DDX5 expression were identified as independent prognostic factors (Table S2). Taken together, these results indicated that DDX5 was overexpressed in highly proliferative NSCLC and might serve as an independent prognostic biomarker for NSCLC.
Figure 2.

Expression of DDX5 is correlated with Ki67 and poor prognosis in patients with non-small-cell lung cancer (NSCLC). (a) Immunohistochemical staining of DDX5 and Ki67 in tumor specimens from lung cancer patients with squamous cell carcinoma and adenocarcinoma. (b) Spearman’s correlation analysis of DDX5 and Ki67 expression in NSCLC samples. (c) Kaplan–Meier analysis of overall survival in 120 patients with surgically resected NSCLC. Low DDX5 protein expression was associated with significantly decreased overall survival.
DDX5 promoted proliferation of NSCLC cells in vitro
To further investigate the biological role of DDX5 expression in NSCLC, squamous cell carcinoma cell line H520 and adenocarcinoma cell line A549 were established to stably overexpress DDX5 (Fig.3a). Absorbance assays showed an approximately 1.6-fold increase in the number of DDX5-overexpressing cells as compared with the control cells after 3 days of culture (Fig.3b). In addition, enhanced cell proliferation by DDX5 upregulation was also observed in EdU incorporation and colony formation assays (Fig.3c,d). Conversely, downregulation of DDX5 significantly slowed down the growth of the cells (Fig.4a,b). Consistently, depletion of endogenous DDX5 in H520 and A549 cells also caused significant inhibition of their DNA synthesis and colony formation ability, as indicated by reduction in EdU-positive cells and colony numbers (Fig.4c,d). Taken together, these results suggested that DDX5 was involved in promoting proliferation of NSCLC cells.
Figure 3.

Overexpression of DDX5 stimulates non-small-cell lung cancer cell proliferation. (a) Squamous carcinoma H520 cells and adenocarcinoma A549 cells were stably infected with DDX5 or vector. The expression of DDX5 was analyzed by Western blot. (b) Cell growth curves as determined by MTT assay. (c) Percentage of EdU-positive cells determined by EdU incorporation assay (lower panel) and representative images of EdU staining (upper panel). (d) Effects of upregulated DDX5 on the clonogenic ability of H520 and A549 cells. Representative images are shown in the upper panel and the number of colonies illustrated in the lower panel. *P < 0.05.
Figure 4.

Downregulation of DDX5 inhibits non-small-cell lung cancer cell proliferation. (a) H520 and A549 cells were stably infected with RNAi-DDX5 or RNAi-Vector. The expression of DDX5 was analyzed by Western blot. (b) Cell growth curves as determined by MTT assay. (c) Percentage of EdU-positive cells determined by EdU incorporation assay (lower panel) and representative images of EdU staining (upper panel). (d) Effects of downregulation of DDX5 on the clonogenic ability of H520 and A549 cells. *P < 0.05.
DDX5 enhanced tumorigenesis of NSCLC cells in vivo
To confirm the pro-proliferative role of DDX5 in vivo, we established a xenograft model using H520 cells stably infected with DDX5 or DDX5-RNAi, as well as the vectors. As shown in Figure5, DDX5 overexpression resulted in significantly larger tumor size and tumor mass compared with the vector control group; DDX5 downregulation significantly decreased the tumor size and tumor mass relative to the shRNA vector control group. Moreover, IHC staining revealed that tumors formed by cells infected with DDX5 showed a significant increase expression of cyclin D1 and Ki67. In contrast, tumors formed by cells infected with DDX5-RNAi displayed a sharply decreased expression of cyclin D1 and Ki67. Collectively, these data further confirmed that DDX5 had a key role in promoting the proliferation and tumorigenesis of NSCLC cells.
Figure 5.
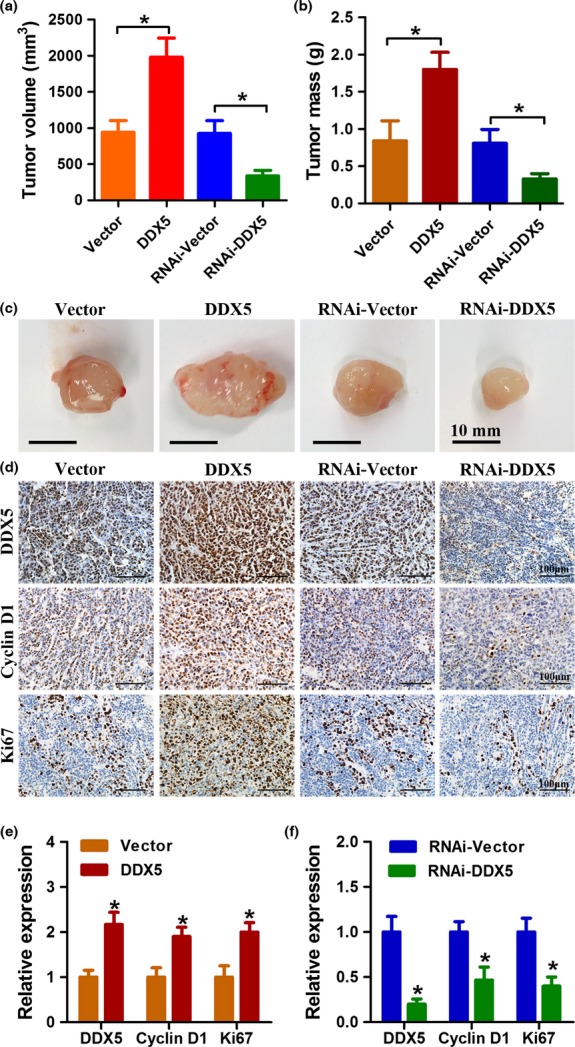
DDX5 promotes non-small-cell lung cancer cell growth in vivo. (a, b) H520 cells were stably infected with the indicated lentivirus and injected s.c. into BALB/c-nude mice. Four weeks after injection, xenografts were removed. Tumor volume and tumor weight were determined. (c) Representative images of xenografts from different groups. (d–f) Immunohistochemical analysis of the expression of DDX5, Ki67, and cyclin D1 in different groups of xenografts. *P < 0.05.
DDX5 promoted NSCLC cell proliferation by activating β-catenin signaling pathway
As DDX5 was reported to participate in the β-catenin signaling pathway in regulating biological behaviors of cancer cells,10,14 we examined whether DDX5-induced cell proliferation in NSCLC was mediated by this signaling pathway. Co-immunoprecipitation assay indicated a direct physical interaction between DDX5 and β-catenin in both H520 and A549 cells (Fig.6a). We also found that both transcriptional and translational levels of β-catenin target genes cyclin D1 and c-Myc were upregulated by DDX5 overexpression, but suppressed by DDX5 silencing, compared with that in control cells (Figs6b,c,S1). Interestingly, we observed that nuclear accumulation of β-catenin was significantly increased (or decreased) by DDX5 overexpression (or downregulation), whereas the expression of total β-catenin was not affected (Figs6c,d,S1). Moreover, the dual luciferase reporter assay revealed that β-catenin/TCF and cyclin D1 promoter activity were significantly increased in the DDX5-overexpressing NSCLC cells, but decreased in the DDX5 silencing cells (Fig.6e,f). These data collectively suggested that DDX5 might promote NSCLC cell proliferation by activating the β-catenin signaling pathway.
Figure 6.

DDX5 promotes β-catenin nuclear accumulation and activates cyclin D1 and c-Myc transcription. (a) Co-immunoprecipitation (IP) analysis of DDX5 and β-catenin in H520 and A549 non-small-cell lung cancer (NSCLC) cells. β-catenin and DDX5 can be reciprocally co-immunoprecipitated by their antibodies. Whole cell lysate was probed for input. Bead lanes contain the protein G-conjugated Sepharose beads used during the immunoprecipitation without the protein input. WB, Western blot. (b) Real-time PCR analysis of the relative expression of cyclin D1 and c-Myc mRNA in NSCLC cells. (c) Western blot analysis of the protein expression of DDX5, (total or nuclear) β-catenin, cyclin D1, and c-Myc. (d) Immunofluorescence analysis of β-catenin in DDX5 or vector-infected NSCLC cells. White arrows indicate nuclear localization of β-catenin. (e, f) Indicated cells transfected with pGL3-cyclinD1 (firefly), TOPflash (TOP) or FOPflash (FOP), and pRL-TK (Renilla) plasmids were subjected to dual luciferase reporter assays 48 h after transfection. Reporter activity was normalized by Renilla luciferase activity. *P < 0.05.
Inhibition of β-catenin signal abrogated DDX5-induced cell proliferation
We further investigated whether inhibiting β-catenin could block DDX5-induced cyclin D1 and c-Myc expression and cell proliferation. Western blot and real-time PCR analysis revealed that siRNA-mediated β-catenin inhibition drastically abolished DDX5-induced cyclin D1 and c-Myc expression (Fig.7a,b). Accordingly, DDX5-enhanced β-catenin/TCF and cyclin D1 promoter activity was also suppressed by β-catenin downregulation (Fig.7c,d). Interestingly, silencing of β-catenin also moderately reduced the expression of DDX5, indicating that DDX5 might also serve as a downstream effector of β-catenin (Fig.7b). Moreover, DDX5-stimulated cell proliferation was also abrogated upon β-catenin inhibition, as revealed by the EdU incorporation assay (Fig.7e). Taken together, these results supported the hypothesis that DDX5-induced proliferation of NSCLC cells was dependent on the β-catenin signaling pathway.
Figure 7.
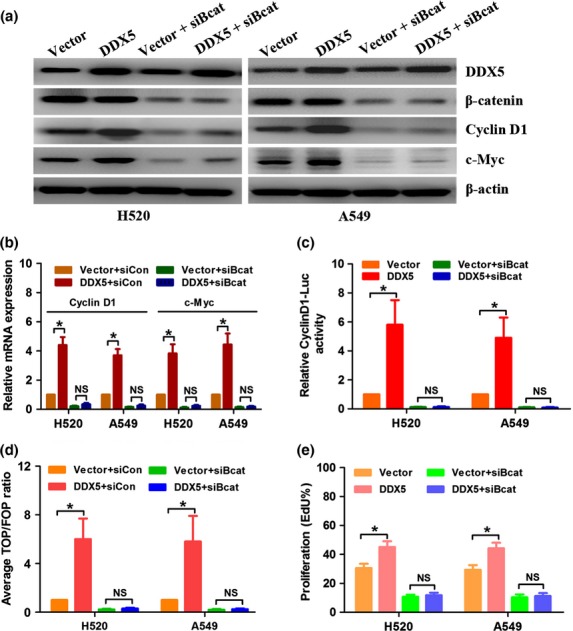
Suppression of β-catenin abrogates DDX5-induced cyclin D1 and c-Myc expression and cell proliferation. (a) Real-time PCR analysis of the expression of cyclin D1 and c-Myc. DDX5-infected H520 and A549 cells were subjected to transfection of siRNA targeting β-catenin (siBcat) or control siRNA (siCon) for 48 h. (b) Western blot analysis of the expression of DDX5, β-catenin, cyclin D1, and c-Myc. (c) Indicated cells were transfected with siBcat or siCon plus pGL3-cyclinD1 (firefly) and pRL-TK (Renilla) for 48 h and subjected to dual luciferase reporter assays. Relative cyclinD1-Luc activity was normalized by Renilla luciferase activity. (d) Indicated cells were transfected with siBcat or siCon plus TOPflash (TOP) or FOPflash (FOP) and pRL-TK (Renilla) plasmids for 48 h and subjected to dual luciferase reporter assays. Reporter activity was normalized by Renilla luciferase activity. (e) EdU incorporation assay revealed that inhibition of β-catenin abrogated DDX5-induced cell proliferation. *P < 0.05. NS, not significant.
Expression of DDX5 correlated with cyclin D1 in clinical NSCLC samples
To confirm whether the results we obtained from NSCLC cell lines could also be obtained in the clinical setting, we examined the expression of cyclin D1 in 120 NSCLC specimens by IHC staining (Fig.8a). Spearman’s correlation analysis revealed that the expression of DDX5 and cyclin D1 showed a significant positive correlation (r = 0.457, P < 0.001; Fig.8b). These results further corroborated our in vitro findings.
Figure 8.
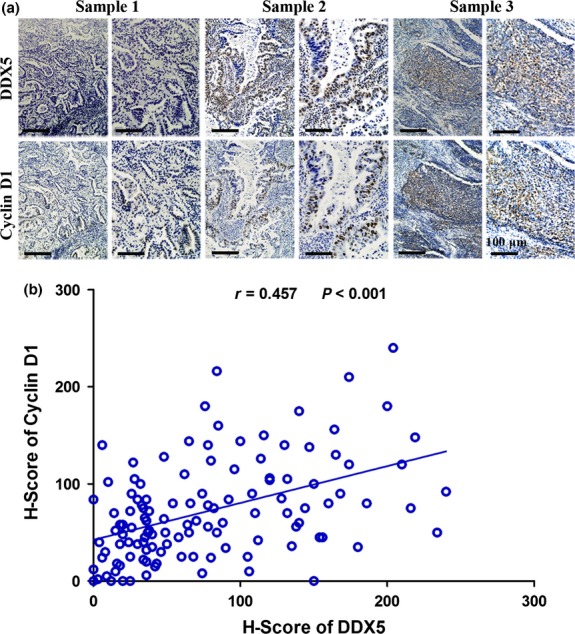
DDX5 expression is positively correlated with cyclin D1 expression in human non-small-cell lung cancer tissues. (a) Representative images of immunohistochemical staining of DDX5 and cyclin D1 in cancer tissues. (b) Correlation analysis of immunohistochemistry scores of DDX5 and cyclin D1.
Discussion
In this study, we found that DDX5 was overexpressed in NSCLC tissues compared with the matched ANTs. In addition, overexpression of DDX5 was significantly correlated with the progression and poor prognosis of NSCLC. We also observed that DDX5 upregulation promoted the proliferation of NSCLC cells in vitro and the growth of NSCLC xenografts in vivo. Conversely, downregulation of DDX5 suppressed the proliferation and tumorigenesis of NSCLC cells. Furthermore, we showed that DDX5 promotes proliferation and tumorigenesis of NSCLC cells by activating the β-catenin signaling pathway. Therefore, our results pointed at DDX5 to be a novel important effector of NSCLC progression.
As a multifunctional protein, dysregulated DDX5 is frequently detected in many malignancies, although, the expression profile, prognostic value, and functional role of DDX5 depend on the cellular context.5,6 DDX5 is reported to be overexpressed in breast cancer, prostate cancer, colorectal cancer, glioma, and leukemia as compared with their normal counterparts, and contributes to proliferation and progression of these tumors.5,6 Overexpression of DDX5 is also associated with poor overall survival in glioma.19 In accordance with these reports, we also detected increased DDX5 protein expression in NSCLC tissues compared with the paired ANTs. Moreover, overexpression of DDX5 is correlated with unfavorable clinicopathological features (e.g. high Ki67 index) and poor OS of patients with NSCLC. The pro-proliferative role of DDX5 inferred from our clinical sample study was also verified by in vitro and in vivo experiments.
β-catenin is implicated in growth and metastasis of various cancers, including NSCLC, by modulating both cell adhesion and gene transcription.20 The β-catenin signaling pathway is one of the most studied mechanisms that mediate DDX5-induced cancer progression. Shin et al.21 first reported that DDX5 and its homologous protein DDX17 promoted colon cancer cell proliferation by forming complexes with β-catenin and activating the transcription of target pro-oncogenes, including c-Myc, cyclin D1, c-Jun, and fra-1. Yang et al.14 revealed that DDX5 mediated platelet-derived growth factor-induced colon cancer cell proliferation and EMT. DDX5 was also implicated in breast cancer progression by maintaining a positive feedback loop with β-catenin/transcription factor 4 signaling.10 Given the above-mentioned reports, we were prompted to investigate the role of this signaling in DDX5-enhanced NSCLC proliferation. As expected, DDX5 exerted pro-proliferative effects by promoting β-catenin nuclear accumulation and co-activating its downstream effector cyclin D1 and c-Myc. This idea was further supported by results showing that inhibiting β-catenin drastically abrogated DDX5-induced cyclin D1 and c-Myc expression and proliferation in NSCLC cells. Moreover, the expression of DDX5 and cyclin D1 in NSCLC patients’ samples displayed a strong positive correlation, indicating a key role of the β-catenin signaling pathway in mediating DDX5-induced NSCLC cell proliferation.
The data herein suggest that DDX5 functions as an oncogenic factor in NSCLC by activating cell proliferation and tumorigenesis through the β-catenin signaling pathway. However, the exact model of the interaction between DDX5 and β-catenin requires further investigation. Moreover, whether elevated DDX5 contributes to other malignant properties of NSCLC cells, such as EMT and metastasis, also remains to be determined. We are currently testing this notion by conducting relevant experiments both in vitro and in vivo.
In conclusion, our study has identified DDX5 as a novel prognostic biomarker and a potential therapeutic target in patients with NSCLC, which may contribute to the development of novel anticancer therapies.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant No. 81070062).
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Relative quantification of the Western blots in H520 (a) and A549 (b) cells based on densitometry by ImageJ.
Table S1. Association between DDX5 expression and clinicopathological features of non-small-cell lung cancer patients.
Table S2. Univariate and multivariate Cox regression analyses of overall survival.
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–46. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn L. Advances in the treatment of non-small cell lung cancer. J Natl Compr Canc Netw. 2014;12:764–7. doi: 10.6004/jnccn.2014.0185. [DOI] [PubMed] [Google Scholar]
- Fuller-Pace FV. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim Biophys Acta. 2013;1829:756–63. doi: 10.1016/j.bbagrm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Fuller-Pace FV, Moore HC. RNA helicases p68 and p72: multifunctional proteins with important implications for cancer development. Future Oncol. 2011;7:239–51. doi: 10.2217/fon.11.1. [DOI] [PubMed] [Google Scholar]
- Dai TY, Cao L, Yang ZC, et al. P68 RNA helicase as a molecular target for cancer therapy. J Exp Clin Cancer Res. 2014;33:64. doi: 10.1186/s13046-014-0064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GJ, Nicol SM, Wilson BJ, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005;24:543–53. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Rid R, Maier CJ, et al. DDX5 is a multifunctional co-activator of steroid hormone receptors. Mol Cell Endocrinol. 2012;361:80–91. doi: 10.1016/j.mce.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Clark EL, Coulson A, Dalgliesh C, et al. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938–46. doi: 10.1158/0008-5472.CAN-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guturi K, Sarkar M, Bhowmik A, Das N, Ghosh M. DEAD-box protein p68 is regulated by ss-catenin/transcription factor 4 to maintain a positive feedback loop in control of breast cancer progression. Breast Cancer Res. 2014;16:496. doi: 10.1186/s13058-014-0496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Thakur MK. Interaction of estrogen receptor-alpha transactivation domain with nuclear proteins of mouse brain: p68 RNA helicase shows age- and sex-specific change. J Neurosci Res. 2009;87:1323–8. doi: 10.1002/jnr.21948. [DOI] [PubMed] [Google Scholar]
- Yang L, Lin C, Zhao S, Wang H, Liu ZR. Phosphorylation of p68 RNA helicase plays a role in platelet-derived growth factor-induced cell proliferation by upregulating cyclin D1 and c-Myc expression. J Biol Chem. 2007;282:16811–9. doi: 10.1074/jbc.M610488200. [DOI] [PubMed] [Google Scholar]
- Wang H, Gao X, Yang JJ, Liu ZR. Interaction between p68 RNA helicase and Ca2+-calmodulin promotes cell migration and metastasis. Nat Commun. 2013;4:1354. doi: 10.1038/ncomms2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127:139–55. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Wang D, Huang J, Hu Z. RNA helicase DDX5 regulates microRNA expression and contributes to cytoskeletal reorganization in basal breast cancer cells. Mol Cell Proteomics. 2012;11:M111.011932. doi: 10.1074/mcp.M111.011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek A, Luo W, Krasnitz A, Hicks J, Powers RS, Stillman B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2012;2:812–25. doi: 10.1158/2159-8290.CD-12-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth A, Cortis J, Soltermann A, et al. Tumour cell proliferation (Ki-67) in non-small cell lung cancer: a critical reappraisal of its prognostic role. Br J Cancer. 2014;111:1222–9. doi: 10.1038/bjc.2014.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Yi X, Liu W, et al. MTDH mediates trastuzumab resistance in HER2 positive breast cancer by decreasing PTEN expression through an NFkappaB-dependent pathway. BMC Cancer. 2014;14:869. doi: 10.1186/1471-2407-14-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Jiao Z, Li R, Yue H, Chen L. p68 RNA helicase promotes glioma cell proliferation in vitro and in vivo via direct regulation of NF-kappaB transcription factor p50. Neuro-oncology. 2012;14:1116–24. doi: 10.1093/neuonc/nos131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:djt356. doi: 10.1093/jnci/djt356. [DOI] [PubMed] [Google Scholar]
- Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–8. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Relative quantification of the Western blots in H520 (a) and A549 (b) cells based on densitometry by ImageJ.
Table S1. Association between DDX5 expression and clinicopathological features of non-small-cell lung cancer patients.
Table S2. Univariate and multivariate Cox regression analyses of overall survival.