

Abstract
Hematopoietic pre-B cell leukemia transcription factor interacting protein (HPIP) has been shown to play an important role in the development and progression of some cancers. However, the role of HPIP in gastric cancer (GC) is unclear. Here, we show that HPIP is upregulated in most GC patients and promotes GC cell proliferation, migration, and invasion. In GC patients, HPIP positively associates with tumor size and nodal metastasis, and negatively associates with tumor differentiation. Hematopoietic pre-B cell leukemia transcription factor interacting protein increases GC cell proliferation through activation of G1/S and G2/M cell cycle transitions, accompanied by a marked increase of the positive cell cycle regulators, including cyclin D1, cyclin A, and cyclin B1. Hematopoietic pre-B cell leukemia transcription factor interacting protein enhances GC cell migration and invasion, and modulates epithelial–mesenchymal transition, which plays a key role in cancer cell migration and invasion. These data underscore the critical role of HPIP in GC cell proliferation and progression and suggest that HPIP inhibition may be a useful therapeutic strategy for GC treatment.
Keywords: Cell growth, EMT, gastric cancer, HPIP, migration and invasion
Gastric cancer (GC) is one of the most common malignant tumor types in the world,1 According to global cancer statistics, GC is the fourth most common malignancy and the second leading cause of cancer death worldwide.2,3 In China, the incidence of gastric cancer ranks third among all malignancies and its mortality rate is 26.3 per 100 000 people.4,5 Thus, investigation of genetic alterations underlying GC tumorigenesis and progression is essential to individual treatment of GC. Although it has been established that gastric tumor progression is associated with a multistep process involving the activation of oncogenes and the inactivation of tumor suppressor genes,6,7 our understanding of the molecular mechanisms underlying the development and progression of GC remains limited.
Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP), also known as pre-B-cell leukemia homeobox-interacting protein 1, was identified by a yeast two-hybrid screen using a human hematopoietic cDNA-based library.8 The HPIP protein has been shown to be involved in organogenesis and tumorigenesis.9,10 It is overexpressed in breast infiltrative ductal carcinoma, astrocytoma, and oral squamous cell carcinoma, and promotes cancer cell proliferation, adhesion, and migration.11–13 Our recent studies showed that HPIP is overexpressed in liver cancer and promotes hepatoma cell proliferation through activation of G2/M cell cycle transition.14 However, the role of HPIP in gastric cancer is unclear.
In this study, we show that expression of HPIP is upregulated in GC tissues compared with matched non-cancerous tissues and, more importantly, HPIP expression associates with some important clinicopathologic factors in GC patients. Hematopoietic pre-B-cell leukemia transcription factor interacting protein promotes GC cell proliferation through activation of G1/S and G2/M cell cycle transitions, accompanied by alteration of expression of important cell cycle regulators. Moreover, HPIP enhances GC cell migration and invasion, and induces GC cell epithelial–mesenchymal transition (EMT), a critical player in regulating cancer cell invasive phenotype.
Materials and Methods
Immunohistochemical analysis
Gastric cancer and adjacent non-cancerous tissues were obtained from Shandong Cancer Hospital and Institute (Jinan, China), with approval and supervision by the Research Ethics Committee of Shandong Cancer Hospital and Institute. Written informed consent was obtained from all patients prior to operation. The immunohistochemical procedure was carried out as described previously.15 Briefly, antigen retrieval was carried out using microwave treatment, and sections were then incubated with rabbit anti-HPIP antibody (Proteintech, Chicago, IL, USA) at a dilution of 1/100. Bound primary antibodies were detected by the addition of biotinylated goat anti-rabbit secondary antibody and streptavidin–HRP (Zymed Laboratories, South San Francisco, CA, USA). The color was visualized with 3,3′-diaminobenzidine (Sigma, St Louis, MO, USA). The samples were counterstained with hematoxylin. Purified IgG from normal rabbit serum (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a negative control. The results were evaluated independently by two pathologists blinded to the origin of the specimen. The widely accepted German semiquantitative scoring system in considering the staining intensity and area extent was used: 0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining. In addition, the percentage of staining was given a score: 0, <5%; 1, 5–25%; 2, 25–50%; 3, 51–75%; and 4, >75%. The two scores mentioned above were multiplied as the final score. For HPIP, we defined a score of 0–2 as negative and 3–12 as positive.
Plasmids, cell lines, and reagents
MGC803 (well differentiated adenocarcinoma), SGC7901 (moderately differentiated adenocarcinoma), and BGC823 (poorly differentiated adenocarcinoma) were purchased from Cell Resource Centre of the Chinese Academy of Sciences (Shanghai, China). MKN-1 (well differentiated adenosquamous carcinoma) was provided by the Riken BRC Cell Bank (Tsukubashi, Japan). Stable cell lines overexpressing HPIP were obtained by lentiviral transduction using pCDH plasmid (System Biosciences, Mountain View, CA, USA). Stable HPIP knockdown cell lines were produced by inserting HPIP shRNA fragments into the lentiviral vector pSIH-H1-Puro (System Biosciences). The sequence of HPIP shRNA has been described previously.16 Lentivirus was made by transfection of the 293T producer cell line with the lentiviral vector and packing vector mix (System Biosciences). Lentivirus was harvested 48 h later, and added to GC cells. Stable cell lines expressing HPIP or HPIP shRNA were selected with puromycin for 48 h after infection. Individual clones were screened by immunoblot with anti-HPIP. Similar results were obtained with pooled clones. Anti-E-cadherin and anti-N-cadherin were obtained from BD Biosciences, San Jose, CA, USA; Anti-vimentin was purchased from Cell Signaling Technology, Danvers, MA, USA, anti-HPIP was gained from Proteintech, and anti-cyclin A, anti-cyclin B1, anti-cyclin D1, and anti-GAPDH were purchased from Santa Cruz Biotechnology.
Cell growth and colony formation assays
Anchorage-dependent cell growth was determined by the CCK-8 Kit (Dojindo Laboratories, Kumamoto, JAPAN) according to the protocol of previous work.17 For colony formation assay, transfected cells were seeded in 6-well plates at 2000 cells per well. Two weeks later, colonies were fixed with 4% paraformaldehyde and stained with crystal violet for 30 min. The number of colonies with diameters of more than 1.5 mm was scored.
Cell cycle analysis
Cell cycle analysis was carried out using flow cytometry as described previously.18 Briefly, cells were fixed in 70% ethanol for approximately 18 h, washed with PBS, and treated with RNase A (0.2 mg/mL) in PBS. Propidium iodide was then added to the cell suspension. Samples were determined by a FACSCalibur Flow Cytometer (Becton Dickinson, New Jersey, USA).
Cell migration and invasion assays
Wound healing assays were carried out to examine cell migration.19 Briefly, transfected cells grown in 6-well plates as confluent monolayers were mechanically scratched using a 1-mL pipette tip to create the wound. Cells were washed with PBS to remove the debris and were grown for 24 h to allow wound healing. Photographs were taken under an inverted phase-contrast fluorescence microscope (Olympus, Tokyo, Japan). Finally, the wound healing rates were calculated, and compared to the width at 0 h. Cell invasion was assessed with Matrigel (BD Biosciences) coated on the upper surface of the Transwell chamber (Corning, NY, USA) as described previously.20 Cells invaded through the Matrigel membrane were fixed with 4% paraformaldehyde and stained with crystal violet. The number of invaded cells was counted in five randomly selected microscopic fields and photographed.
Statistical analysis
All in vitro experiments were carried out in triplicate and repeated three times. The difference of HPIP expression between gastric cancers and normal tissues was determined by Mann–Whitney U-test. Statistical significance in cell proliferation, migration, and invasion assays among constructs was assessed by two-tailed Student’s t-test. The spss 17.0 statistical software Chicago, USA package was used to carry out the statistical analyses. P < 0.05 was considered statistically significant.
Results
Overexpression of HPIP in GC patients
We examined the expression of HPIP by IHC on tissues consisting of 103 pairs of human gastric tumors and adjacent normal gastric mucosa; HPIP was localized mainly in the cytoplasm. According to HPIP scores, HPIP expression was significantly overexpressed in GC patients (P = 1.92 × 10−8) (Fig. 1a–c). For paired tumor and normal tissues, in 71.9% (74/103) of patients, the expression levels of HPIP in tumors were higher than those in adjacent normal tissues. In 15.5% (16/103) of patients, the cancers expressed lower levels of HPIP than normal tissues, whereas in 12.6% (13/103) of patients, the expression levels of HPIP in tumors was relatively equal to that in normal tissues. The specificity of anti-HPIP antibody was verified by immunohistochemical staining of GC tissues incubated with anti-HPIP preincubated with its antigen (Fig. 1d) and immunoblotting of lysates from BCG823 and SGC7901 GC cells infected with HPIP shRNA (Fig. 1e).
Figure 1.

Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) expression is upregulated in gastric cancer patients. (a) Representative immunohistochemical staining of HPIP protein in gastric carcinoma tissue (left) and matched adjacent normal gastric tissue (right). Case 1, poor to moderately differentiated adenocarcinoma. Case 2, moderately to well differentiated adenocarcinoma. Scale bar = 100 μm. The HPIP expression scores are displayed in box-and-whisker plots (b) and bar charts (c) and compared (Mann–Whitney U-test). (d) Immunohistochemical staining of a representative gastric cancer sample incubated with normal IgG or anti-HPIP. To validate antibody specificity, the anti-HPIP was pre-incubated with purified recombinant GST-tagged HPIP (GST-HPIP) protein or GST for 1 h prior to applying to tissue. Original magnification, ×20. Scale bar = 100 μm. (e) Immunoblot analysis of lysates from BCG823 (left panel) or SGC7901 (right panel) cells infected with lentivirus carrying control shRNA or HPIP shRNA using antibodies specific for anti-HPIP. MW, molecular weight. (f) Representative immunohistochemical staining of HPIP protein in gastric carcinoma tissue with different tumor differentiation: well differentiated (left), moderately differentiated (middle), and poorly differentiated (right). Original magnification, ×10. Scale bar: 100 μm. (g) Box plot of HPIP expression in 103 GC tissues with different differentiation status. Data were analyzed by one-way anova test with Games–Howell’s correction. Horizontal lines represent the median, the bottom and top of the boxes represent the 25th and 75th percentiles, respectively, and the vertical bars represent the range of data. Any outliers are marked with a circle.
Correlation between HPIP and clinical parameters in human GC samples
To further investigate the clinical significance of HPIP, we determined the relationship between HPIP and clinical parameters in 103 GC patients. Pearson’s χ2-test showed that HPIP expression positively associated with tumor size and nodal metastasis, and negatively associated with tumor differentiation (Fig. 1f,g), but did not correlate with age, gender, or histological type (Table1).
Table 1.
Correlations between hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) status and clinicopathologic factors in patients with gastric cancer
| Clinical characteristics | Total cases | HPIP (low) | HPIP (high) | P-value† |
|---|---|---|---|---|
| Age, years | ||||
| ≤50 | 50 | 16 | 34 | 0.672 |
| >50 | 53 | 18 | 35 | |
| Gender | ||||
| Male | 55 | 16 | 39 | 0.406 |
| Female | 48 | 18 | 30 | |
| Tumor size, mm | ||||
| ≤20 | 47 | 23 | 24 | 0.003 |
| >20 | 56 | 11 | 45 | |
| Nodal metastasis | ||||
| Yes | 61 | 10 | 51 | 0.001 |
| No | 42 | 24 | 18 | |
| Histological type | ||||
| Adenocarcinoma | 64 | 25 | 39 | 0.13 |
| Signet-ring cell carcinoma | 39 | 9 | 30 | |
| Differentiation | ||||
| High | 18 | 12 | 6 | 0.003 |
| Moderate | 45 | 13 | 32 | |
| Poor | 40 | 9 | 31 | |
P-values assessed using Pearson’s χ2-test.
Gastric cancer cell proliferation enhanced by HPIP
Next, the effect of HPIP on anchorage-dependent growth of GC cells was examined. All four GC cell lines (BGC823, MGC803, SGC7901, and MKN-1) tested expressed similar levels of endogenous HPIP protein (Fig. 2a). Thus, to investigate the role of HPIP in GC, we chose both BGC823 and SGC7901 cell lines to overexpress and knockdown HPIP. As expected, BGC823 and SGC7901 cells infected with HPIP-expressing lentivirus grew much faster than those infected with empty vector (Fig. 2b), whereas BGC823 and SGC7901 cells infected with HPIP shRNA grew more slowly than those transfected with control shRNA (Fig. 2c). Furthermore, colony formation assays indicated that overexpression of HPIP in BGC823 and SGC7901 cells increased colony number and colony size (Fig. 2d), whereas knockdown of HPIP with HPIP shRNA in BGC823 and SGC7901 cells decreased the colony number and size (Fig. 2e). Taken together, these results indicated that HPIP enhances the proliferation and colony formation of GC cells.
Figure 2.

Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) increases gastric cancer (GC) cell proliferation. (a) Total proteins extracted from the indicated GC cell lines were analyzed by immunoblotting with anti-HPIP. GAPDH was used as a loading control. (b) BCG823 or SGC7901 cells infected with pCDH-HPIP or pCDH empty vector were grown in regular medium and harvested at the indicated times. Cell number was determined by CCK-8 assay. The representative immunoblot with anti-HPIP indicates HPIP expression levels. (c) BCG823 or SGC7901 cells infected with HPIP shRNA or control shRNA were grown and analyzed as in (b). (d) Colony formation assays of BCG823 or SGC7901 cells infected with pCDH-HPIP or pCDH empty vector. (e) Colony formation assays of BCG823 or SGC7901 cells infected with HPIP shRNA or control shRNA. All values shown are mean ± SD of triplicate measurements and were repeated three times with similar results (*P < 0.05 vs empty vector or control shRNA, **P < 0.01 vs empty vector or control shRNA).
G1/S and G2/M transitions in GC cells stimulated by HPIP
To determine how HPIP regulates GC cell proliferation, we tested the effect of HPIP on cell cycle distribution by flow cytometry analysis Fig. 3. Compared with the control cells, HPIP overexpression in BGC823 cells led to a reduction in the proportion of cells in G0/G1 phase (from 54.94 ± 4.32% to 47.51 ± 2.77%) and G2/M phase (from 17.78 ± 4.18% to 12.63 ± 3.49%), but an increase in the proportion of cells in S phase (from 27.29 ± 3.58% to 39.86 ± 5.73%) (Fig. 3a). Conversely, HPIP knockdown in BGC823 cells significantly enhanced the proportion of cells in both G0/G1 (56.62 ± 3.31% to 63.88 ± 4.09%) and G2/M (14.06 ± 1.47% to 19.71 ± 2.25%) phases, which associated with reduced proportion of cells in S phase (29.32 ± 3.24% to 16.41 ± 1.90%) (Fig. 3b). Similar results were obtained in SGC7901 cells (Fig. 3e,g). These results indicate that HPIP stimulates both the G1/S and G2/M transitions in GC cells.
Figure 3.

Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) activates the G1/S and G2/M transitions in gastric cancer cells. (a) Flow cytometry analysis of cell cycle in BCG823 cells infected with pCDH empty vector or pCDH-HPIP. (b) Flow cytometry analysis of cell cycle in BCG823 cells infected with control shRNA or HPIP shRNA. The experiments were repeated three times with similar trends and the image displayed is one of the representative results. (c, d) Representative immunoblot with the indicated antibodies using cell lysates from (a) and (b). (e) Flow cytometry analysis of cell cycle in SGC7901 cells infected with pCDH empty vector or pCDH-HPIP. (f) Flow cytometry analysis of cell cycle in SGC7901 cells infected with control shRNA or HPIP shRNA. (g, h) Representative immunoblot with the indicated antibodies using cell lysates from (e) and (f). All values shown are mean ± SD of triplicate measurements and were repeated three times with similar results.
Expression of G1 and G2 phase-related proteins in GC cells modulated by HPIP
As HPIP modulates cell cycle distribution, we determined the expression of several important cell cycle-related proteins in HPIP knockdown or overexpressing GC cells. Overexpression of HPIP in BGC823 cells enhanced the expression of the G1/S phase markers cyclin D1 and cyclin A as well as the G2/M phase marker cyclin B1 (Fig. 3c). In contrast, HPIP knockdown in BGC823 cells reduced the expression of cyclins D1, A, and B1 (Fig. 3d).
Gastric cancer cell migration and invasion enhanced by HPIP
Wound healing assays were carried out to evaluate the effect of HPIP on GC cell migration. Overexpression of HPIP in BGC823 and SGC7901 cells increased migration ability (Fig. 4a,b), while HPIP knockdown in these cells inhibited migration ability (Fig. 4c,d). The Transwell invasion assay showed that HPIP overexpression in BGC823 cells increased the number of invaded cells (Fig. 4e), whereas HPIP knockdown in BGC823 cells decreased the number of invaded cells (Fig. 4f).
Figure 4.

Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) increases gastric cancer cell migration and invasion. (a, b) Wound healing assays of BCG823 (a) or SGC7901 (b) cells infected with pCDH-HPIP or pCDH empty vector. All values shown are mean ± SD of triplicate measurements and were repeated three times with similar results. The image displayed is one of the representative results. Scale bar = 100 μm. (c, d) Wound healing assays of BCG823 (c) or SGC7901 (d) cells infected with control shRNA or HPIP shRNA. Cells were analyzed as in (a) and (b). (e, f) BGC823 cells infected with pCDH-HPIP (e) or HPIP shRNA (f) were assessed by Matrigel invasion chamber. Invasive cells were fixed and stained with crystal violet. The number of invaded cells was counted. Scale bar = 100 μm. All values shown are mean ± SD of triplicate measurements and were repeated three times with similar results. *P < 0.05 versus pCDH empty vector or control shRNA.
Epithelial–mesenchymal transition of GC cells promoted by HPIP
As EMT is well known to be involved in invasion and metastasis of cancer cells,21–23 we tested the effects of HPIP on EMT in GC cells. Consistent with the results of HPIP modulation of GC cell migration and invasion, HPIP overexpression induced EMT, with the activation of morphologic changes from a polarized epithelial phenotype to an elongated fibroblastoid phenotype (Fig. 5a). Opposite results were seen in HPIP knockdown cells (Fig. 5b). Furthermore, HPIP overexpression reduced the expression of the epithelial marker E-cadherin and increased that of N-cadherin and vimentin, two mesenchymal markers (Fig. 5a, right panel). Again, opposite results were observed in HPIP knockdown cells (Fig. 5b, right panel).
Figure 5.
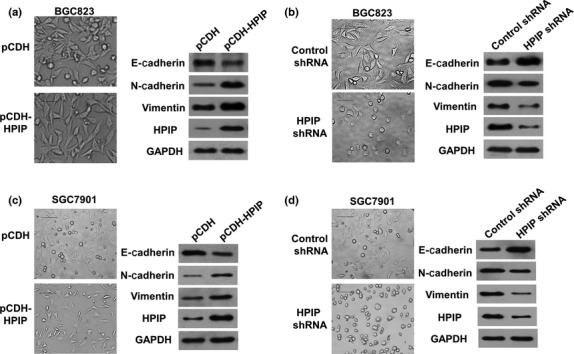
Hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) promotes epithelial–mesenchymal transition of gastric cancer cells. BGC823 cells were infected with pCDH-HPIP (a) or HPIP shRNA (b). Representative morphologic changes are shown in the photographs. Scale bar = 100 μm. Whole cell extracts from (a) and (b) were used for representative immunoblots with the indicated antibodies.
Knockdown of HPIP inhibits GC cell growth in mouse model
Finally, we investigated the effect of HPIP on GC cell growth in a mouse model. BGC823 cells stably infected with HPIP shRNA lentivirus or empty vector were inoculated s.c. in the dorsal skin fold of each nude mouse. As expected, knockdown of HPIP significantly suppressed the GC growth in nude mice (Fig. 6a), compared with the control shRNA group. In addition, the BGC823 tumors with HPIP shRNA showed decreased expression of HPIP, cyclins D1, A, and B1, and N-cadherin, and increased expression of E-cadherin (Fig. 6b).
Figure 6.
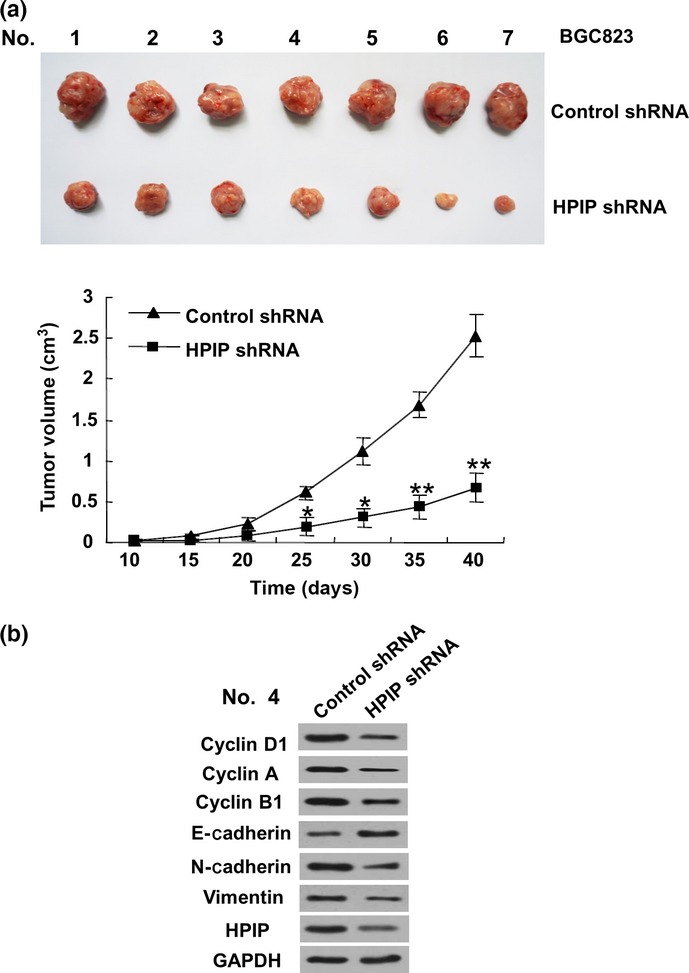
Knockdown of hematopoietic pre-B-cell leukemia transcription factor interacting protein (HPIP) suppresses gastric cancer cell growth in nude mice. (a) BGC823 cells stably infected with HPIP shRNA or control shRNA cells were injected into nude mice. At the indicated times, tumors were measured with Vernier calipers (mean ± SD; n = 7). **P < 0.01 versus corresponding control shRNA. (b) Immunoblot analysis of representative excised tumor from (a).
Discussion
Gastric cancer is one of the leading causes of cancer death worldwide, with hundreds of thousands of newly diagnosed cases as well as related deaths per year.21 Improved understanding of this deadly cancer at the basic molecular level is particularly needed. Although HPIP has been shown to be involved in the development and progression of several cancers,10–14 the importance of HPIP in gastric cancer is still unclear. In this study, we showed for the first time a fundamental role of HPIP as an oncogene in gastric carcinoma. First, HPIP is overexpressed in gastric carcinoma, and associates with several important clinicopathologic factors, such as tumor size, nodal metastasis status, and tumor differentiation of GC patients, indicating the clinical significance of HPIP in GC. Second, HPIP increases both cell growth and colony formation of GC cells. It activates GC cell cycle progression, accompanied by changes in expression of important cell cycle regulators. Finally, HPIP promotes GC cell migration and invasion with increased EMT. These findings suggest that HPIP may contribute to gastric carcinogenesis and metastasis.
Hematopoietic pre-B cell leukemia transcription factor interacting protein was shown to be overexpressed in patients with breast infiltrative ductal carcinoma. The protein physically interacts with estrogen receptor α and functionally modulates estrogen signaling. Thus, HPIP promotes breast cancer cell growth through enhanced estrogen signaling. It also interacts with focal adhesion (FA) complex containing FA kinase (FAK), stimulates FAK, and promotes breast cancer cell migration in a FAK-dependent manner.11 Hematopoietic pre-B cell leukemia transcription factor interacting protein is also overexpressed in liver cancer patients. It regulates hepatoma cell proliferation through G2/M cell cycle activation and enhances anchorage-dependent and -independent growth of human liver cancer cells. Overexpression of HPIP increases hepatoma cell proliferation, migration, and invasion, and promotes EMT through regulation of mTOR signaling.14,16 Similar to upregulation of HPIP expression in breast and liver cancer patients, upregulation of HPIP expression has also been reported in glioma and oral squamous cell carcinoma.12,13 Knockdown of HPIP reduces glioma cell viability and motility through rearrangements of the actin cytoskeleton. Our study showed that HPIP is overexpressed in GC patients, and associates with tumor size, nodal metastasis, and differentiation in GC patients. Similar to HPIP overexpression in liver cancer cells, HPIP overexpression in GC cells enhances cell proliferation, migration, and invasion, and induces EMT. Combined with previous findings, our data establish HPIP as an important oncogene. Thus, HPIP inhibition may be a useful therapeutic strategy for GC treatment. Those HPIP-specific siRNAs or microRNAs (miRNAs) that target HPIP, such as miR-148a, which has been shown to inhibit HPIP expression, might be developed to treat HPIP-overexpressing GC patients by repressing HPIP expression. These molecules might be chemically modified, and might be produced in the form of liposome formulations, nanoparticles, and targeting moieties. Small chemical molecules might also be developed to inhibit GC cell growth by reducing the HPIP protein level.
One of the most important factors predicting outcome of patients with GC is the status of lymph node metastasis. Several staging systems for GC are based on this parameter, such as the staging system of the American Joint Committee on Cancer and the International Union Against Cancer published in 1997.22,23 In addition to lymph node metastasis, differentiation of GC has been more and more emphasized in assessing tumor progression and outcome of GC patients. Several research groups show that the 5-year survival rate is higher in GC patients with intestinal type of cancer (differentiated type) than in those with diffuse type of cancer (undifferentiated type), and the 5-year survival rate is higher in GC patients with expanding type cancer (differentiated type) than in those with infiltrative type cancer (undifferentiated type).24,25 Adachi et al.26 further demonstrated that differentiation is one of the independent prognostic factors among the pathologic variables of GC using multivariate analysis. Although HPIP has been shown to be upregulated in breast infiltrative ductal carcinoma, colorectal cancer,27 liver cancer, astrocytoma, and oral squamous cell carcinoma (OSCC), correlation of HPIP expression with lymph node metastasis and differentiation is unclear in all of these cancers except OSCC. There is no significant association between HPIP and lymph node metastasis and differentiation in OSCC. In our study, we show that HPIP expression is correlated with differentiation negatively and with nodal metastasis positively. As differentiation and nodal metastasis are two of the most important pathologic variables in cancer patients, our data indicate that HPIP may play a key role in predicting poor clinical outcome in GC patients.
Metastasis is the major cause for the high mortality rate of gastric cancer. Epithelial–mesenchymal transition is a fundamental process in embryonic development and is considered as an important step leading to tumor invasion and metastasis.28–30 The most observed character of EMT is that cells take on a spindle-like morphology and experience loss of epithelial cell markers, such as E-cadherin, and gain of mesenchymal markers, including N-cadherin and vimentin.31–34 Our study showed that HPIP regulates morphologic change in GC cells, accompanied by alterations in the expression of epithelial and mesenchymal markers. It will be interesting to investigate how HPIP modulates GC cell EMT and metastasis in more detail.
We previously showed that HPIP is a direct target of miRNA-148a, which was downregulated in liver cancer patients. MicroRNA-148a has also been shown to be significantly downregulated in GC tissues compared to adjacent normal gastric tissues. In addition, downregulated miR-148a in GC significantly correlates with TNM stage, lymph node metastasis and poor clinical outcome. Functionally, overexpression of miR-148a suppresses GC cell invasion and metastasis in vitro and lung metastasis formation in vivo.35 As in GC patients, miR-148a expression is downregulated (previous study) and HPIP expression is upregulated (this study); it could be expected that expression of miR-148a might be inversely correlated with HPIP expression in GC.
Acknowledgments
The work was supported by the China Major State Basic Research Development Program (Grant Nos. 2011CB504202 and 2012CB945100), the National Natural Science Foundation (Grant Nos. 81330053, 81272913, 81472589, 81402345, and 31100604), and the Beijing Nova Program (Grant No. Z141102001814055).
Disclosure Statement
The authors have no conflict of interest.
References
- Kang C, Song JJ, Lee J, Kim MY. Epigenetics: an emerging player in gastric cancer. World J Gastroenterol. 2014;20:6433–47. doi: 10.3748/wjg.v20.i21.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Awais D, Siegel CA, Higgins PD. Modelling dysplasia detection in ulcerative colitis: clinicalimplications of surveillance intensity. Gut. 2009;58:1498–503. doi: 10.1136/gut.2008.169714. [DOI] [PubMed] [Google Scholar]
- Yeoh KG. How do we improve outcomes for gastric cancer? J Gastroenterol Hepatol. 2007;22:970–2. doi: 10.1111/j.1440-1746.2007.04956.x. [DOI] [PubMed] [Google Scholar]
- Sun XD, Mu R, Zhou YS, et al. Analysis of mortality rate of stomach cancer and its trend in twenty years in China. Zhonghua Zhongliu Zazhi. 2004;26:4–9. [PubMed] [Google Scholar]
- Röcken C, Warneke V. Molecular pathology of gastric cancer. Pathologe. 2012;33:235–40. doi: 10.1007/s00292-012-1634-4. [DOI] [PubMed] [Google Scholar]
- Cervantes A, Rodríguez Braun E, Pérez Fidalgo A, Chirivella González I. Molecular biology of gastric cancer. Clin Transl Oncol. 2007;9:208–15. doi: 10.1007/s12094-007-0041-4. [DOI] [PubMed] [Google Scholar]
- Abramovich C, Shen WF, Pineault N, et al. Functional cloning and characterization of a novel nonhomeodomain protein that inhibits the binding of PBX1-HOX complexes to DNA. J Biol Chem. 2000;275:26172–7. doi: 10.1074/jbc.M001323200. [DOI] [PubMed] [Google Scholar]
- Manavathi B, Acconcia F, Rayala S, Kumar R. An inherent role of microtubule network in the action of nuclear receptor. Proc Natl Acad Sci USA. 2006;103:15981–6. doi: 10.1073/pnas.0607445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang Z, Zhang H, et al. The estrogen receptor-interacting protein HPIP increases estrogen-responsive gene expression through activation of MAPK and AKT. Biochim Biophys Acta. 2008;1783:1220–8. doi: 10.1016/j.bbamcr.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Bugide S, David D, Nair A, et al. Hematopoietic PBX-interacting protein (HPIP) is overexpressed in breast infiltrative ductal carcinoma and regulates cell adhesion and migration through modulation of focal adhesion dynamics. Oncogene. 2014 doi: 10.1038/onc.2014.389. doi: 10.1038/onc.2014.389. [DOI] [PubMed] [Google Scholar]
- van Vuurden DG, Aronica E, Hulleman E, et al. Pre-B-cell leukemia homeobox interacting protein 1 is overexpressed in astrocytoma and promotes tumor cell growth and migration. Neuro Oncol. 2014;16:946–59. doi: 10.1093/neuonc/not308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Irie T, Tanaka J, et al. Potential role of hematopoietic pre-B-cell leukemia transcription factor-interacting protein in oral carcinogenesis. J Oral Pathol Med. 2014;24:1–11. doi: 10.1111/jop.12210. [DOI] [PubMed] [Google Scholar]
- Xu X, Jiang C, Wang S, et al. HPIP is upregulated in liver cancer and promotes hepatoma cell proliferation via activation of G2/M transition. IUBMB Life. 2013;65:873–82. doi: 10.1002/iub.1202. [DOI] [PubMed] [Google Scholar]
- Pan X, Zhou T, Tai Y, et al. Elevated expression of CUEDC2 protein confers endocrine resistance in breast cancer. Nat Med. 2011;17:708–14. doi: 10.1038/nm.2369. [DOI] [PubMed] [Google Scholar]
- Xu X, Fan Z, Kang L, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123:630–45. doi: 10.1172/JCI64265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Xu X, Kang L, et al. miR-30a suppresses breast cancer cell proliferation and migration by targeting Eya2. Biochem Biophys Res Commun. 2014;445:314–9. doi: 10.1016/j.bbrc.2014.01.174. [DOI] [PubMed] [Google Scholar]
- Wang C, Tao W, Ni S, et al. Tumor-suppressive microRNA-145 induces growth arrest by targeting SENP1 in human prostate cancer cells. Cancer Sci. 2015;4:375–82. doi: 10.1111/cas.12626. doi: 10.1111/cas.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Cheng L, Wang Y, et al. The RNA-binding protein RBPMS1 represses AP-1 signaling and regulates breast cancer cell proliferation and migration. Biochim Biophys Acta. 2015;1853:1–13. doi: 10.1016/j.bbamcr.2014.09.022. [DOI] [PubMed] [Google Scholar]
- He XJ, Jiang XT, Ma YY, et al. REG4 contributes to the invasiveness of pancreatic cancer by upregulating MMP-7 and MMP-9. Cancer Sci. 2012;103:2082–91. doi: 10.1111/cas.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kodera Y, Yamamura Y, Shimizu Y, et al. The number of metastatic lymph nodes: a promising prognostic determinant for gastric carcinoma in the latest edition of the TNM classification. J Am Coll Surg. 1998;187:597–603. doi: 10.1016/s1072-7515(98)00229-4. [DOI] [PubMed] [Google Scholar]
- Ichikura T, Tomimatsu S, Uefuji K, et al. Evaluation of the new American Joint Committee on Cancer/International Union Against Cancer classification of lymph node metastasis from gastric carcinoma in comparison with the Japanese classification. Cancer. 1999;86:553–8. doi: 10.1002/(sici)1097-0142(19990815)86:4<553::aid-cncr2>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Noda S, Soejima K, Inokuchi K. Clinicopathological analysis of the intestinal type and diffuse type of gastric carcinoma. Jpn J Surg. 1980;10:277–83. doi: 10.1007/BF02468788. [DOI] [PubMed] [Google Scholar]
- Riberio MM, Sarmento JA, Simoes MAS, et al. Prognostic significance of Lauren and Ming classifications and other pathologic parameters in gastric carcinoma. Cancer. 1981;47:780–4. doi: 10.1002/1097-0142(19810215)47:4<780::aid-cncr2820470424>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Adachi Y, Yasuda K, Inomata M, et al. Pathology and prognosis of gastric carcinoma: well versus poorly differentiated type. Cancer. 2000;89:1418–24. [PubMed] [Google Scholar]
- Feng Y, Xu X, Zhang Y, et al. HPIP is upregulated in colorectal cancer and regulates colorectal cancer cell proliferation, apoptosis and invasion. Sci Rep. 2015;5:9429. doi: 10.1038/srep09429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenrieder V, Hendler SF, Boeck W, et al. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001;61:4222–8. [PubMed] [Google Scholar]
- Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial- mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang H, Liu J, et al. MiR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem Biophys Res Commun. 2012;417:1100–5. doi: 10.1016/j.bbrc.2011.12.121. [DOI] [PubMed] [Google Scholar]
- Yan W, Fu Y, Tian D, et al. PI3 kinase/Akt signaling mediates epithelial-mesenchymal transition in hypoxic hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009;382:631–6. doi: 10.1016/j.bbrc.2009.03.088. [DOI] [PubMed] [Google Scholar]
- Xia J, Guo X, Yan J, et al. The role of miR-148a in gastric cancer. J Cancer Res Clin Oncol. 2014;140:1451–6. doi: 10.1007/s00432-014-1649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]