

Abstract
Cellular causes of resistance and limited drug distribution within solid tumors limit therapeutic efficacy of anticancer drugs. Acidic endosomes in cancer cells mediate autophagy, which facilitates survival of stressed cells, and may contribute to drug resistance. Basic drugs (e.g. doxorubicin) are sequestered in acidic endosomes, thereby diverting drugs from their target DNA and decreasing penetration to distal cells. Proton pump inhibitors (PPIs) may raise endosomal pH, with potential to improve drug efficacy and distribution in solid tumors. We determined the effects of the PPI lansoprazole to modify the activity of doxorubicin. To gain insight into its mechanisms, we studied the effects of lansoprazole on endosomal pH, and on the spatial distribution of doxorubicin, and of biomarkers reflecting its activity, using in vitro and murine models. Lansoprazole showed concentration-dependent effects to raise endosomal pH and to inhibit endosomal sequestration of doxorubicin in cultured tumor cells. Lansoprazole was not toxic to cancer cells but potentiated the cytotoxicity of doxorubicin and enhanced its penetration through multilayered cell cultures. In solid tumors, lansoprazole improved the distribution of doxorubicin but also increased expression of biomarkers of drug activity throughout the tumor. Combined treatment with lansoprazole and doxorubicin was more effective in delaying tumor growth as compared to either agent alone. Together, lansoprazole enhances the therapeutic effects of doxorubicin both by improving its distribution and increasing its activity in solid tumors. Use of PPIs to improve drug distribution and to inhibit autophagy represents a promising strategy to enhance the effectiveness of anticancer drugs in solid tumors.
Keywords: Doxorubicin, drug distribution, lansoprazole, pharmacodynamic markers, solid tumor
The development of intrinsic or acquired drug resistance remains the major obstacle to clinical efficacy of cancer chemotherapy. Mechanistic studies of poor responsiveness of tumor cells to cytotoxic agents have hitherto concentrated on a multitude of molecular changes occurring in individual cells with emphasis on stable genetic changes that may be induced or selected for by therapy.1 Anticancer drugs access solid tumors through the blood stream and must penetrate through multiple cell layers of the extravascular space in order to reach all of the target tumor cells. An essential but rather under-studied factor that may jeopardize the effectiveness of anticancer drugs (including molecular targeted agents) against solid tumors is the limited capability of drugs to localize to all constituent tumor cells in sufficient concentrations to induce lethality; if drugs only reach a proportion of their target cells in low concentration, their overall effects will be compromised, regardless of their mode of action or potency.2 Accumulating evidence has indicated limited perivascular distribution of the auto-fluorescent anticancer drugs doxorubicin, mitoxantrone, and topotecan in solid tumors, in contrast to uniform distribution in most normal tissues other than brain.(3,4) We have also used markers of DNA damage and apoptosis to show that a panel of other clinically used anticancer drugs, including docetaxel and melphalan, display limited spatial distribution from tumor blood vessels.5 Inadequate drug delivery through tumor tissues results in preferential killing of cells adjacent to patent blood vessels both as a consequence of exposure to higher concentrations of cytotoxic agents, but also because these cells proliferate rapidly and are therefore sensitive to most anticancer drugs. In contrast, there is limited activity against slower-proliferating cells residing far away from the vasculature that receive little or no drug exposure.(6,7) Slowly proliferating distal cells are likely to survive and repopulate the tumor following chemotherapy treatment.8
Irregularly organized vascular architecture and lack of lymphatic drainage in solid tumors leads to formation of regions with deficiency in the supply of oxygen and nutrients as well as build-up of abundant metabolic breakdown by-products, predominantly lactic and carbonic acid, establishing an acidic extracellular microenvironment (pHe 6.5–6.9).9 When weakly basic anticancer drugs encounter an acidic extracellular milieu, they are quickly ionized and converted into positively charged protonated species; this attenuates cellular uptake because charged forms are relatively membrane impermeant.10 Even if basic compounds diffuse through the cell membrane, sequestration and accumulation within acidic vesicles (e.g. recycling endosomes and lysosomes) will occur, leaving a small fraction of drugs available to attack nuclear DNA and exert antitumor effects.11
The pH gradients in cancer cells are finely controlled by different ion/proton pumps including the vacuolar-type H+-ATPase (V-ATPase), whose elevated expression is positively linked with metastatic potential and multiple drug resistance (MDR) in multiple tumor types.(12,13) The V-ATPases shuttle free protons across the membranes of a wide array of cytoplasmic compartments and are critical for the regulation and maintenance of endosomal pH.14 A class of H+/K+-ATPase inhibitors known as proton pump inhibitors (PPIs) hinders acidification of parietal cells in the wall of the stomach and these drugs are used widely as first-line treatment of peptic diseases. These pro-drugs also inhibit the action of V-ATPases, although higher concentrations are required than to inhibit gastric acid secretion.15 Proton pump inhibitors accumulate selectively in acidic spaces and have been shown to raise both extracellular pH and the pH of acidic organelles by blocking the formation and outward flux of H+.16 Pretreatment with PPIs may alter intracellular drug distribution by attenuating the trapping of weak bases in acidic organelles, which will allow more drugs both to enter the nucleus and cause cytotoxicity, and to exit the cell and penetrate to more distant cells from vasculature. Because of their effects to raise endosomal pH, PPIs also inhibit autophagy, a process that may facilitate survival of stressed or damaged cells through recycling of cellular molecules that are broken down within the acidic compartments of cells.(17,18) Investigations from our and other laboratories indicate that induction of autophagy is a mechanism of resistance to anticancer drugs that can be suppressed by PPIs and other agents to improve therapeutic efficacy.(18–21)
We postulate that manipulation of pH within the extracellular space and within intracellular organelles of tumor cells may have considerable potential to improve the outcome of some chemotherapeutic drugs for solid tumors by improving drug distribution. Our laboratory and others have shown that the PPIs omeprazole, esomeprazole, and pantoprazole were able to improve drug penetration, enhance effectiveness of chemotherapy, and restore drug sensitivity in in vitro and in vivo models, although there is uncertainty about their major mechanism(s) of action.(18,22–25) In the present study, we investigate the impact of the PPI lansoprazole on endosomal acidity, cytotoxicity, and penetration of the weakly basic drug doxorubicin using in vitro models of dispersed cells and multilayered cell cultures (MCC). To obtain further insight into mechanisms operative in solid tumors, we use quantitative immunohistochemistry to determine the influence of lansoprazole both to modify the spatial distribution of doxorubicin (evaluated by its auto-fluorescence) and of biomarkers (γH2AX and cleaved caspase-3 or -6) that reflect cytotoxic activity.
Materials and Methods
Drugs and reagents
Doxorubicin (Pharmacia, Missisauga, ON, Canada) was obtained from the Princess Margaret Cancer Centre (Toronto, Canada) pharmacy as a solution at a concentration of 2 mg/mL. Lansoprazole was purchased from Sigma (St. Louis, MO, USA) as a dry powder, dissolved in 100% ethanol and diluted with 0.9% saline to the appropriate concentrations immediately before administration. DiOC7 was purchased from AnaSpec (San Jose, CA, USA) and a stock solution (2.5 mg/mL) was made by dissolving in DMSO. The stock was 1:10 diluted in PBS and 10% Solutol HS 15 (BASF, Ludwigshafen, Germany) shortly prior to use. Rabbit anti-human γH2AX and cleaved caspase-3 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-human cleaved caspase-6 antibody was purchased from Novus Biologicals (Oakville, ON, Canada). Cy3-conjugated goat anti-rabbit IgG secondary antibody was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Cell culture and tumor models
Mouse mammary carcinoma EMT6 cells were provided by Dr. Peter Twentyman (Medical Research Council, Cambridge, UK) and human mammary adenocarcinoma MCF7 cells were purchased from ATCC (Manassas, VA, USA). These two cell lines were chosen because they represent two different species and their properties allow reproducible growth of MCC with uniform thickness.26 Both cell lines were maintained as monolayers in α-MEM media supplemented with 10% FBS (HyClone, Logan, UT, USA) and antibiotics at 37°C in a humidified atmosphere of 95% air and 5% CO2. Routine tests to exclude mycoplasma and characterize the origin of cells (short tandem repeat analysis) were carried out every 6 months. All in vitro experiments were carried out using exponentially growing cells and were repeated at least twice in triplicate.
To generate MCF7 xenografts, 4–6-week-old female athymic nude mice (Jackson Laboratory, Bar Harbor, ME, USA) with implanted 17β-estradiol tablets (60 day release; Innovative Research of America, Sarasota, FL, USA) in the dorsal interscapular region were injected s.c. in both flanks with 5 × 106 MCF7 cells per side. To generate EMT6 tumors, female syngeneic Balb/C mice were injected with 1 × 106 EMT6 cells. Mice were acclimatized in the animal colony under specific pathogen-free conditions for 1 week before experimentation and received filtered water and sterile rodent food ad libitum. There were five to six mice per treatment group (∼10–12 tumors) and each experiment was repeated twice. All procedures were carried out following approval of the Institutional Animal Care Committee in accordance with the Canadian Council on Animal Care guidelines.
Measurement of endosomal pH
Briefly, 1 × 106 EMT6 and MCF7 cells were exposed to various doses of lansoprazole in the presence or absence of doxorubicin (1.8 μM). Cells were subsequently incubated for 3 h with fresh media containing dextran–fluorescein–tetramethylrhodamine 10 000 MW, anionic (FITC/TMR-dextran; Molecular Probes, Eugene, OR, USA), which was taken up into endosomes, followed by culture in media for 2 h. Changes in endosomal pH were determined by measuring fluorescence on a Beckman Coulter Mississauga, ON, Canada) EPICS Elite flow cytometer equipped with an argon–ion laser emitting at 488 nm. The argon laser was adopted to excite FITC and TMR with emission at the wavelength of 525 nm (pH-dependent) and 575 nm (pH-independent), respectively. Calibration of fluorescence measurements was carried out using a standard curve generated by the fluorescence spectrum of the ionophore nigericin (Sigma) in buffers with known pH values, as described previously.23 A standard curve representing the relationship between FITC fluorescence emission ratio and pH was established. The mean fluorescence intensity was calculated for each sample and the auto-fluorescence from unlabeled controls was subtracted. At least 10 000 events were acquired and analyzed per sample.
Distribution of doxorubicin in cells
In brief, EMT6 and MCF7 cells seeded on chambered cover glass slides were pretreated with 500 μM lansoprazole for 2 h, followed by incubation for 1 h in media containing 3.6 μM doxorubicin for 2 h. After washing three times with PBS, intracellular doxorubicin fluorescent signals were visualized by using a Zeiss (Oberkochen, Germany) Axiovert 200M fluorescence inverted microscope and were captured with a Roper Scientific (Tucson, AZ, USA) CoolSnap HQ charge-coupled device camera. Doxorubicin fluorescence was excited with an argon laser at 514 nm and the emission was collected at 488 nm.
Colony-forming assay
Single cell suspensions were treated in glass polyshell vials with desired final concentrations of lansoprazole ranging from 100 μM to 1 mM alone or in combination with 1.8 μM doxorubicin. The vials were incubated in a water bath at 37°C, magnetically stirred, and gassed with 95% air and 5% CO2. Samples were removed as a function of time up to 5 h later, centrifuged, suspended in fresh media, and seeded in serial dilutions in six-well plates. After 10 days of incubation, cells were fixed with 4% paraformaldehyde and stained with methylene blue and colonies consisting of >50 cells were counted. Clonogenic survival was expressed as the ratio of the plating efficiencies (total colony number/cells plated) of treated to untreated cells and was plotted using a logarithmic scale against drug concentration.
Doxorubicin penetration in MCCs
Approximately 2 × 105 EMT6 or MCF7 cells were seeded on collagen-coated microporous Teflon membranes of culture plate inserts (Millipore Billerica, MA, USA) and given 6 h to attach to the membrane. The membranes were submerged in a large reservoir of stirred α-MEM media to allow feeding from both sides of the membrane and incubated at 37°C for periods of ∼6–8 days; this resulted in the formation of MCCs composed of ∼5 × 106 cells. The MCCs were examined under a light microscope to ensure uniform thickness (∼150−200 μm) prior to use in experiments. The MCCs were incubated in either media alone or media containing 250 μM lansoprazole for 2 h. Solutions containing 3.6 μM doxorubicin were prepared in FBS-free media and mixed well with 1% agar in a 1:1 ratio. Agar prevents convective motion from influencing penetration properties but does not inhibit drug transport. A volume of 0.5 mL drug solution was added to one side of MCCs. The membranes were floated in glass polyshell vials containing fresh media and incubated at 37°C in an atmosphere of 95% air and 5% CO2. After exposure to doxorubicin for 1 h, MCCs were removed and snap-frozen in liquid nitrogen and 10-μm sections were imaged using an Olympus (Tokyo, Japan) Upright BX50 microscope and a 100-W HBO mercury light source equipped with 530–560 nm excitation and 573–647 nm emission wavelength filter sets.
We used immunohistochemistry to investigate the spatial penetration of doxorubicin through MCCs over time. We first converted the top layer of each MCC into an 8-bit (255 arbitrary units) white binary image with a defined intensity of 255. Next, each image was overlaid with the corresponding field of view displaying drug intensity, resulting in an 8-bit black and white image with the drug source identified by an intensity of 255 (white) and drug fluorescence as a gray-scale ranging from 0 to 254 units. Areas of interest were selected from each section and were on average 1600 × 1600 μm (0.4 μm2/pixel). Areas of necrosis and staining artifact were excluded. To minimize noise from auto-fluorescence, a minimal threshold for detection was determined for each MCC. The pixel intensity and distance to the drug source for all pixels within each area of interest above the threshold were assessed with a sophisticated algorithm as previously described.3 Doxorubicin fluorescence intensity relative to background was averaged over all pixels at any given distance from the drug source and plotted as a function of that distance.
Imaging and quantification of doxorubicin distribution in solid tumors
Animals with EMT6 or MCF7 s.c. tumors were treated with saline, doxorubicin alone, or lansoprazole before doxorubicin once tumors reached a diameter of 8–12 mm. Doxorubicin was given as a single i.v. injection (25 mg/kg) to facilitate detection and quantification of its auto-fluorescence. Lansoprazole (200 mg/kg) was given i.p. 2 h before doxorubicin treatment. The perfusion marker DiOC7 (1 mg/kg) was injected i.v. 1 min prior to tumor extraction to allow visualization of functional blood vessels. Mice were killed 10 min after doxorubicin injection because it was documented that doxorubicin is maximally distributed in solid tumors between 10 min and 3 h after injection.3 The removed tumors were immediately embedded into Tissue-Tek OCT (Sakura Finetek, Torrance, CA, USA), snap-frozen in liquid nitrogen and stored at −80°C prior to tissue sectioning. For each tumor, 10-μm cryostat sections were cut at three different levels approximately 100 μm apart and mounted on slides. A minimum of 10 tumors was analyzed per treatment group.
Doxorubicin and DiOC7 fluorescence was detected using an Olympus Upright BX50 microscope camera. Signals were recorded with a Photometrics (Tucson, AZ, USA) CoolSNAP HQ2 camera using the Cy3 (530–560 nm excitation / 573–647 nm emission) and FITC (490 nm excitation/525 nm emission) filter sets. Images were tiled using an automated stage to ensure that the distribution of doxorubicin was analyzed for the whole tissue section. Composite images were generated by overlaying those for doxorubicin and blood vessels using Media Cybernetics Image Pro Plus Software (version 6.0; Silver Spring, MD, USA). Doxorubicin staining was converted to an 8-bit gray-scale with fluorescence intensities ranging from 1 to 254, while images displaying DiOC7 staining were converted to an 8-bit (255 arbitrary units) black and white binary image so that blood vessels were represented by an intensity of 255. Tumor edges, regions of necrosis, and any artifacts due to processing were excluded from analysis. Background fluorescence, representing the lowest fluorescence intensity within the tumor tissue in each experiment, was subtracted. Quantitative analysis of overlayed images was carried out as noted earlier for MCCs and as per our previous study3 by applying a customized algorithm to generate drug-intensity distributions in relation to distance from the nearest blood vessel. A cut-off of 60 μm was used to minimize interference from neighboring blood vessels that are out of the plane of the section.
Imaging and quantitative analysis of biomarker distributions in solid tumors
Tumor sections were first imaged for DiOC7 using an FITC filter set. Subsequently, tissue sections were washed, blocked, and stained for biomarkers of drug effect (DNA damage marker, γH2AX; markers of apoptosis, cleaved caspase-3 or caspase-6) with appropriate antibodies and imaged using the Cy3 filter set. As MCF7 cells have a 47-bp deletion within the exon 3 of caspase-3,27 we chose to study the expression of cleaved caspase-6 that processes caspase-8 and caspase-10 to trigger apoptosis. γH2AX was expressed within 10 min after exposure to drugs, and activated caspase-3 or caspase-6 at 24 h after treatment.5 A novel protocol was used to quantitatively analyze composite images consisting of the vascular distance map and biomarker signals.28 Briefly, binarized DIOC7 images were first built and applied to create a distance map such that each pixel is represented by its distance to the nearest functional blood vessel in the section. Biomarker distributions were then evaluated by generating binary masks that were combined with the blood vessel distance map using a pixel-by-pixel logical “AND” function to form an overlaid image with distance measurements corresponding only to biomarker-positive pixels. A minimum threshold for detection was determined for each section according to average background readings from unstained regions. Data were presented graphically as the mean frequency of biomarker-positive pixels at each distance from the nearest blood vessel in the section. A cut-off of 60 μm was used to minimize noise from neighboring blood vessels.
Growth delay experiments
Mice with s.c. EMT6 tumors or MCF7 xenografts in both flanks were randomly divided into four groups of five to six mice each and drug treatment commenced when the average tumor diameter was in the range of 5–8 mm. Animals were treated with multiple doses of either saline, doxorubicin alone (6 mg/kg i.v.), lansoprazole alone (150 mg/kg i.p.), or lansoprazole 2 h before doxorubicin weekly for a total of 3 consecutive weeks. Every 2–3 days, two perpendicular diameters of tumors were measured using calipers and body weight was also determined to record possible toxicity due to treatments. Tumor volume (V) was estimated by the following formula: V = A × B2/2, wherein A and B are the longest and the shortest diameters, respectively. Measurements were carried out until tumors reached a maximum diameter of 1.2 cm or began to ulcerate, when mice were killed humanely. All mice were coded using ear tags and randomized to avoid bias in measurements. Due to the fast-growing nature of EMT6 cells in vivo, growth delay studies in these tumors had to be completed within 2 weeks in order to prevent ulceration and overgrown tumors.
Statistical analysis
All data quantification and statistical analyses were carried out with spss 13.0 software (SPSS, Chicago, IL, USA). A one-way anova, followed by Tukey’s post-hoc test, was done to determine statistical differences between experimental groups. Data are presented as the mean ± SEM and all experiments were independently repeated at least twice in triplicate. P-values < 0.05 were considered statistically significant.
Results
Lansoprazole increased endosomal pH and impacted on doxorubicin uptake in breast tumor cells
As depicted in Figure1(a), lansoprazole treatment led to a concentration-dependent increase in endosomal pH in both EMT6 and MCF7 cells; elevation in endosomal pH was consistently observed in the two cell lines with concentrations of lansoprazole above 250 μM. Exposure to 1 mM lansoprazole raised endosomal pH in EMT6 and MCF7 cells by a maximum of approximately 1–2 pH units (Fig.1a). Photomicrographs of fluorescence in MCF7 and EMT6 cells exposed to doxorubicin alone show that doxorubicin was not only localized in the nucleus, where it is known to intercalate with nuclear DNA and induce DNA damage, but also was sequestered in a punctate pattern in the cytoplasm (Fig.1b, panels i,iii, arrows). In contrast, treatment with lansoprazole (500 μM) lowered the amount of doxorubicin fluorescence present in the perinuclear acidic compartments to undetectable levels, whereas it was retained within the nucleus (Fig.1b, panels ii,iv).
Figure 1.
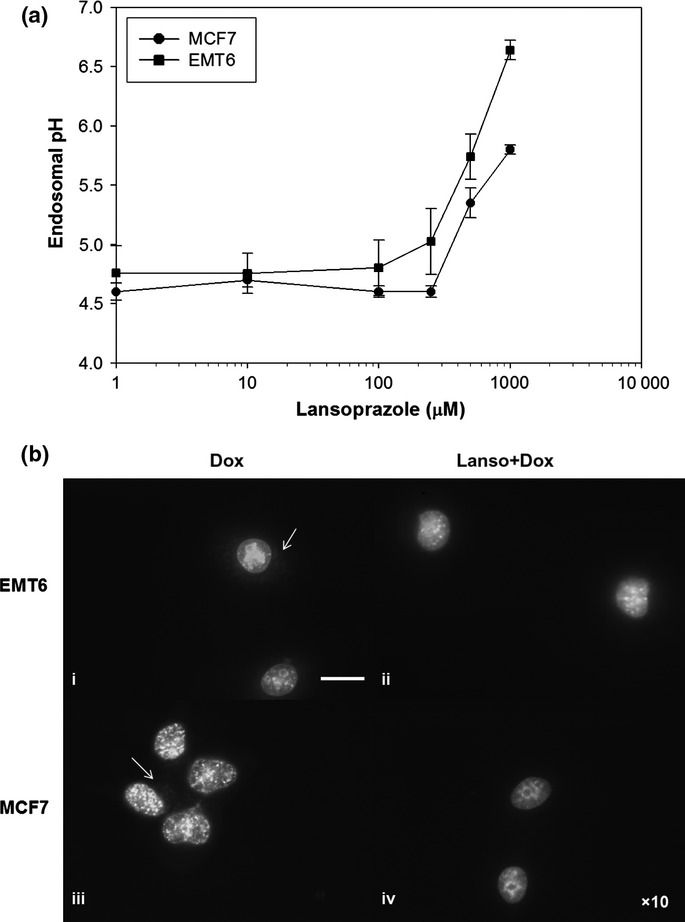
(a) Relationship between endosomal pH and lansoprazole concentration in EMT6 and MCF7 cells. Data are presented as mean ± SEM. (b) Lansoprazole (Lanso) pretreatment prevented accumulation of doxorubicin (Dox) into acidic compartments and modified its intracellular localization. Fluorescent micrographs showing EMT6 and MCF7 cells treated with 3.6 μM doxorubicin (ii, iv) and 500 μM lansoprazole 2 h before doxorubicin. Arrows indicate doxorubicin in cytoplasmic organelles. Scale bar = 20 μm.
Lansoprazole enhanced cytotoxicity of doxorubicin in vitro
We examined whether lansoprazole pretreatment could sensitize cultured EMT6 and MCF7 cells to doxorubicin using a colony-forming assay. Exposure of EMT6 cells to lansoprazole at concentrations of 100 μM, 250 μM, and 500 μM for up to 5 h was not cytotoxic to cells (Fig.2a). However, there was a marked decrease in cell survival when cells were treated with lansoprazole at the highest dose of 1 mM. Similar cytotoxic effects were also observed following exposure of MCF7 cells to lansoprazole at different time points (Fig.2b). When given in combination with 1.8 μM doxorubicin, lansoprazole at concentrations ≥100 μM significantly increased the cytotoxicity of doxorubicin in a dose-dependent manner for both EMT6 and MCF7 cells as compared to doxorubicin alone (P < 0.05; Fig.2c,d).
Figure 2.

Effect of Lansoprazole alone (a, b) or with doxorubicin (c, d) on the cytotoxicity for EMT6 (a, c) and MCF7 (b, d) cells as determined by a clonogenic survival assay. Data are presented as the mean ± SEM.
Lansoprazole pretreatment enhanced doxorubicin penetration through MCCs
We next tested whether lansoprazole enables drugs to have improved penetration through tumor tissue using the MCC model. This model allows tumor cells to be grown in culture with many properties reflecting tumors in vivo, including desmosomes between cells, generation of ECM components, gradients of nutrient concentration, and cell proliferation, and provides a simple and direct means for quantification of drug transport through a tissue-like environment.29 In EMT6- and MCF7-derived MCCs exposed only to doxorubicin, doxorubicin fluorescence was predominantly accumulated in superficial layers at the edge of MCCs at 2 h after application of the drug (Fig.3a,b), while cells situated at deeper layers from the drug source appeared to have minimal drug uptake. When MCCs were pretreated with 250 μM lansoprazole, there was a marked increase in doxorubicin penetration in MCCs grown from both tumor types and doxorubicin fluorescence was detectable in cells more distal from the source of drug as compared to controls (Fig.3c,d). This was supported by quantitative analysis of doxorubicin fluorescence as a function of depth in the MCCs (Fig.3e,f).
Figure 3.

Penetration of doxorubicin (Dox) through multilayered cell cultures (MCCs) generated from EMT6 (a, c) and MCF7 (b, d) cells with and without lansoprazole (Lanso) pretreatment. Note the increased doxorubicin (Dox) fluorescence in regions more distal from the source of drug in MCCs that were pretreated with 250 μM lansoprazole (c, d) compared to controls (a, b) at 2 h following drug exposure (3.6 μM doxorubicin). (e, f) Quantitative analysis of doxorubicin as a function of distance from the drug source in EMT6- and MCF7-derived MCCs pretreated with lansoprazole in comparison with controls. Doxorubicin fluorescence intensity was quantified at 2 h after the drug was given as a function of the depth from the top of MCCs. Data are presented as the mean ± SEM. Scale bar = 100 μm.
Treatment with lansoprazole improved doxorubicin distribution in tumors
The potential effects of lansoprazole to modify doxorubicin distribution in relation to functional blood vessels were evaluated in transplanted tumors derived from EMT6 and MCF7 cells. Ten minutes after injection, distribution of doxorubicin in EMT6 and MCF7 tumors treated with doxorubicin alone was poor and was limited to perivascular regions; there was a rapid decline in doxorubicin fluorescence intensity with increasing distance from blood vessels (Fig.4a,d). Pretreatment with lansoprazole led to a substantial increase in doxorubicin fluorescence, particularly in areas situated distant from blood vessels, in both EMT6 and MCF7 tumors compared with those exposed to doxorubicin alone (Fig.4b,e). Quantification of the doxorubicin distribution at 10 min after injection showed steep gradients of reducing doxorubicin fluorescence relative to distance from the nearest functional blood vessel in all tumors (Fig.4c,f). A significantly shallower gradient of reducing doxorubicin intensity was identified in EMT6 tumors pretreated with lansoprazole when compared with tumors treated with doxorubicin alone (P < 0.05; Fig.4c). A similar trend was also seen in MCF7 tumors pretreated with lansoprazole compared to those in the doxorubicin only group, although the difference in drug distribution between the two treatment groups was not statistically significant (Fig.4f).
Figure 4.

Distribution of doxorubicin fluorescence intensity in relation to blood vessels in EMT6- and MCF7-derived tumors. Mice were given either 25 mg/kg doxorubicin alone ((Dox; 10 min) (a, d) or 200 mg/kg lansoprazole (2 h before doxorubicin) and doxorubicin (Dox+Lanso) (b, e) prior to tumor excision. Tumors were imaged and quantified using a customized algorithm to show the mean absolute (background-subtracted) values of doxorubicin fluorescence intensity (blue) as a function of distance to the nearest functional blood vessel (green) (c, f). Scale bar = 100 μm.
Lansoprazole pretreatment improved distribution of pharmacodynamic biomarkers in tumor sections
The distribution of activity of neoplastic agents depends not only on the distribution of native drugs, but also on that of active metabolites and on the distribution of cellular sensitivity to them. We therefore also evaluated the spatial distribution of pharmacodynamic markers of drug effect in relation to the nearest patent functional blood vessel in the tumors. As shown in Figures5 and S1, treatment with saline or lansoprazole alone did not lead to activation of biomarkers. In EMT6 tumors and MCF7 xenografts treated with doxorubicin alone, expression of molecular markers was increased in all regions compared to those treated with saline and lansoprazole alone (P < 0.05; Figs5,S1). Moreover, distributions of induced γH2AX at 10 min as well as cleaved caspase-3 or -6 at 24 h following treatment were largely restricted to regions adjacent to blood vessels and their expression declined with increasing distance from blood vessels, mirroring quite closely the distribution pattern of doxorubicin (Fig.4). Treatment of the two types of tumor with lansoprazole and doxorubicin produced the most profound effects on the biomarkers throughout tumor sections as compared to other treatment groups (P < 0.05). A significant increase in DNA damage and apoptosis was observed in regions both proximal and distal to blood vessels in the combination group compared to chemotherapy alone (Figs5,S1), suggesting that lansoprazole pretreatment promoted both tumor cell sensitivity to doxorubicin and the spatial distribution of drug activity in solid tumors.
Figure 5.

Comparison of distribution of pharmacodynamic biomarkers γH2AX and cleaved caspase-3 or caspase-6 relative to functional blood vessels in EMT6 (a, c) and MCF7-derived (b, d) tumors following treatments: saline (Control), 200 mg/kg lansoprazole (Lansop), 25 mg/kg doxorubicin (Dox), or lansoprazole plus doxorubicin (Lanso+Dox). γH2AX was evaluated at 10 min post-treatment and cleaved caspase-3 and caspase-6 24 h post-treatment. Data are presented as mean ± SEM.
Lansoprazole pretreament inhibited tumor growth in vivo
Mice treated with saline or lansoprazole alone (up to 250 mg/kg i.p.) once weekly for 3 weeks showed a minimal increase in body weight over the period of >21 days. Doxorubicin treatment alone (6 mg/kg i.v.) exerted no significant influence on body weight. Animals treated with 100 mg/kg lansoprazole followed by doxorubicin once a week for 3 weeks had a temporary loss of body weight by ∼5–8% within the first week of treatment, followed by a rapid regain of their original body weight. Combined treatment with lansoprazole and doxorubicin resulted in an average of ∼11% decrease in body weight when lansoprazole was given at 150 mg/kg (data not shown). Combined treatment with doxorubicin and lansoprazole at doses of 200 mg/kg and higher led to a continual loss in body weight over the course of treatment. Therefore, 150 mg/kg was chosen as the maximum tolerated dose when given together with doxorubicin. All animals remained active and alive throughout the experiments.
Lansoprazole alone showed minimal effects on slowing growth of EMT6 and MCF7 tumors in vivo compared to the control arm treated with saline (Fig.6). Doxorubicin treatment alone resulted in modest growth delay of tumors, whereas multiple doses of lansoprazole and doxorubicin led to the greatest delay in the growth of both EMT6 and MCF7 tumors in comparison with control, lansoprazole, and doxorubicin alone treatment groups (Fig.6). The difference was statistically significant between the lansoprazole plus doxorubicin arm and other individual groups (P < 0.05). Compared to MCF7 xenografts growth of EMT6-derived tumors was delayed to a lesser extent with combined lansoprazole and doxorubicin treatment, probably because of their rapid growth (Fig.6).
Figure 6.
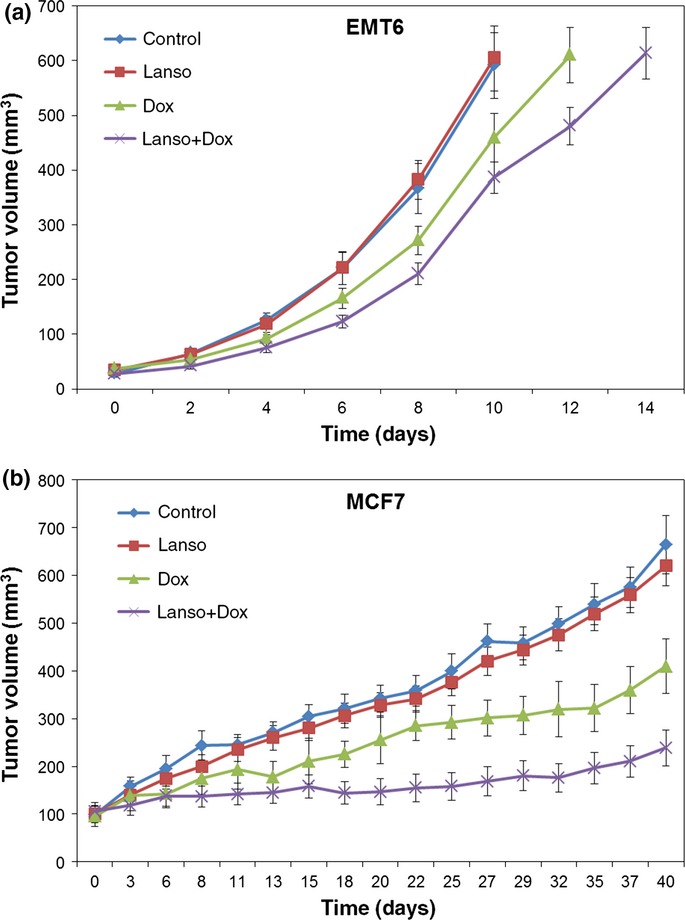
Growth delay of EMT6 tumors (a) and MCF7 xenografts (b) in mice following treatments with saline (Control), 150 mg/kg i.p. lansoprazole (Lanso), 6 mg/kg i.v. doxorubicin (Dox), or the combination of doxorubicin preceded 2 h earlier by lansoprazole (Lanso+Dox) (treatments were given once weekly for 3 weeks). Data are presented as mean ± SEM.
Discussion
The tumor microenvironment is as an integral part of tumor development and many hallmarks of cancer are influenced by stromal components. The acidic microenvironment in solid tumors occurs because of production of lactic and carbonic acids, due to poor vascular perfusion and to high rates of glycolysis and is closely intertwined with tumor initiation and progression; it is also associated with resistance to some anticancer drugs.30 The pH gradient between the cytoplasm and the acidic extracellular space hampers transport of basic drugs into cancer cells, and may also increase elimination of anticancer drugs, such as cisplatin, into the extracellular space by exosomes.(31,32) Once inside tumor cells, sequestration of basic drugs will occur in acidic organelles such as endosomes, thereby inhibiting their access to their molecular target (usually DNA), and limiting drug available for penetration to tumor cells distant from blood vessels. All of these effects may diminish responsiveness of solid tumors to cytotoxic therapy. Acidic endosomes are also important intermediaries in the process of autophagy, which is tightly regulated by autophagy-related genes and their associated enzymes.33 Most evidence suggests that autophagy is a protective and adaptive mechanism, which favors cell survival through degrading and recycling cellular components under stressed conditions (e.g. acidic or hypoxic stress), although autophagy might promote cell death in some tumors.34 There is emerging evidence that upregulation of autophagy may occur following various types of cancer therapy, and that autophagy may be associated with resistance to multiple anticancer drugs.(19–21) Acidification of intracellular compartments in tumor cells is amenable to manipulation by PPIs and other drugs, which might improve the therapeutic index by inhibiting the accumulation of basic cytotoxic agents in acidic vesicles and thereby improving their intratumor distribution, and/or by inhibiting autophagy.
Some PPIs have shown toxicity for cultured tumor cells, while several studies (including the present investigation) have indicated their promising activity to enhance sensitivity to anticancer drugs.(16,30,35) For example, lansoprazole pretreatment sensitized human melanoma cells to suboptimal doses of paclitaxel and combined treatment had a significantly greater efficacy against metastatic melanoma cells, as compared to either drug alone, both in vitro and in vivo.36 Esomeprazole induced rapid death of melanoma and osteosarcoma cells through a caspase-dependent pathway involving early accumulation of reactive oxygen species and induced their susceptibility to cisplatin.25 In addition, PPIs have shown promising activity against a variety of transplanted solid tumors in animal models and have reverted chemoresistance in selective drug-resistant tumors.(18,37–40) Encouraging results have been reported in preliminary studies both in animals with spontaneous tumors and in humans.(25,41,42) For example, high doses of lansoprazole appeared to reverse drug resistance in a cohort of domestic animals with spontaneous chemoresistant neoplasms, leading to a high percentage of responders with minimal toxicity.41 Also, clinical usage of PPIs in a large cohort of patients with head and neck cancer was associated with clinical benefit and was an independent prognostic factor for overall survival.43
The present study and our previous investigations22 provide evidence that PPIs can potentiate therapeutic effectiveness of doxorubicin against solid tumors, in part by increasing its availability for tissue penetration. This might occur through: (i) inhibition of endosomal sequestration of doxorubicin; (ii) inhibition of drug release in exosomes;32 and/or (iii) inhibition of drug extrusion from cells via P-glycoprotein or other MDR-related mechanisms. For example, pantoprazole had the ability to reverse the MDR phenotype of gastric adenocarcinoma cells by downregulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway.44 However, the influence of lansoprazole to enhance doxorubicin distribution in the present study was modest, and a novel aspect of the experimental design was the quantitative analysis of the spatial distribution of doxorubicin itself (determined by the fluorescence of the drug), and of the biomarkers γH2AX and cleaved caspases, which indicate DNA damage and apoptosis, respectively. Lansoprazole had substantial effects on modifying these biomarkers of activity throughout the tumors when given prior to doxorubicin, compared to much smaller effects on modifying the distribution of doxorubicin fluorescence (Fig.4c,f vs. 5). These data are consistent with our previous results showing upregulation of autophagy by the non-basic drug docetaxel in all regions of solid tumors, and its inhibition by the PPI pantoprazole;18 they suggest that lansoprazole may be exerting its therapeutic effects through similar mechanisms.
It has been shown that PPIs are capable of inhibiting autophagy, most likely through inhibition of endosomal acidification and/or of fusion of endosomes with autophagosomes.(18,45) However, a previous study reported time-dependent induction of autophagy in esomeprazole-treated melanoma cells,46 so the influence of PPIs on autophagy may be cell-line dependent. Chloroquine and hydroxychloroquine, two well-known inhibitors of autophagy, have shown effects to enhance chemotherapy in preclinical studies and clinical trials, but their ocular toxicity has restricted clinical application. Novel and safer inhibitors of autophagy are worthy of investigation for their effects to modify treatments with drugs and radiotherapy.
Although our current and previous results show that PPIs raise endosomal pH in cultured cells and can modify the intracellular distribution of basic drugs, the required concentration (>250 μM in the present study) is greater than doses likely to be tolerated in vivo.(47,48) Despite this discrepancy, multiple lines of evidence show activity of these drugs in vivo, including inhibition of autophagy. In solid tumors, the extracellular pH is low in regions distant from patent blood vessels, and acidity increases activity of PPIs. The PPIs might be effective in inhibiting the fusion of endosomes with autophagosomes, as occurs during autophagy, at doses lower than those necessary to raise endosomal pH. In addition, the finding that lower doses of lansoprazole are effective in vivo might be due to the role of the microenvironment and its effects on ion gradients between intracellular compartments and between intracellular and extracellular pH. Our HPLC data have indicated the peak plasma concentrations of pantoprazole in mice treated with 200 mg/kg pantoprazole was ∼300 μM within 1 h and ∼150 μM after 2 h, which were lower than the concentration of pantoprazole (>500 μM) required to raise endosome pH in the in vitro experiments.22 To better understand the mechanism behind lansoprazole’s therapeutic efficacy in vivo, it would be important to examine plasma levels of lansoprazole in mice at various time points.
Our study has several limitations. First, distribution of doxorubicin and markers of drug activity was only assessed in 2D tumor sections, which do not recapitulate completely the 3D features of solid tumors. Second, this analysis was limited to the two selected tumor cell lines and one basic anticancer drug. Ideally our study should be extended to evaluate whether lansoprazole could enhance the antitumor activity of other weak bases (e.g. mitoxantrone and vincristine) and non-basic drugs (e.g. docetaxel, as we reported for pantoprazole) using different spontaneous or orthotopic tumor models. Third, our study did not consider some microenvironmental factors that may affect drug distribution in solid tumors, such as high interstitial fluid pressure (IFP). Elevated IFP occurs due to a dense ECM and high cell density that lead to compression of blood vessels, and to poor lymphatic drainage.(2,9) Provenzano et al.49 showed that targeting hyaluronic acid by the enzymatic agent PEGPH20 normalized IFP and improved delivery of co-administered drugs with a significant inhibition of tumor growth in a pancreatic cancer model.
Promising preclinical results from our group and others led us to undertake a phase I clinical trial of the combination of pantoprazole and doxorubicin, which showed that relatively high doses of pantoprazole could be used in combination with standard dose doxorubicin in patients with advanced solid tumors (NCT01163903).50 We have also initiated an ongoing phase II clinical trial in which pantoprazole is given prior to treatment of men with castrate-resistant prostate cancer with docetaxel (NCT01748500).
In summary, our data show that high-dose lansoprazole inhibited endosomal sequestration of the basic anticancer drug doxorubicin and enhanced doxorubicin cytotoxicity in breast tumor cells. Lansoprazole improved tissue penetration of doxorubicin in MCCs and had a modest effect on improving its spatial distribution in transplanted tumors; lansoprazole had a much larger effect on increasing expression of biomarkers of drug toxicity in all regions of tumors. Multiple doses of lansoprazole prior to delivery of doxorubicin produced a beneficial impact on delaying tumor growth in vivo. These findings endorse the concept that pretreatment with PPIs may be an effective strategy to increase the therapeutic efficacy of chemotherapy in some solid tumors. Multiple clinical trials are ongoing to provide clinical proof of the concept for use of PPIs in the treatment of malignant cancers (www.clinicaltrials.gov).
Acknowledgments
We would like to thank Dr. Krupa Patel for her collaboration in the endosomal pH and drug uptake experiments. We are grateful to all members of the Pathology Research Program and the Advanced Optical Microscopy Facility, University Health Network for providing support with immunohistochemical staining and imaging analysis. This work was supported by a grant from the Canadian Institute for Health Research (MOP-106657).
Disclosure Statement
Ian Tannock received research grants from the Canadian Institute of Health Research for this work. The authors had no conflicts of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Expression of pharmacodynamic biomarkers γH2AX and cleaved caspase-3 or caspase-6 in EMT6 and MCF7 tumors.
References
- Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85:1219–26. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11:8782–8. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Tredan O, Tannock IF. Distribution of the anticancer drugs doxorubicin, mitoxantrone and topotecan in tumors and normal tissues. Cancer Chemother Pharmacol. 2013;72:127–38. doi: 10.1007/s00280-013-2176-z. [DOI] [PubMed] [Google Scholar]
- Saggar JK, Fung AS, Patel KJ, Tannock IF. Use of molecular biomarkers to quantify the spatial distribution of effects of anticancer drugs in solid tumors. Mol Cancer Ther. 2013a;12:542–52. doi: 10.1158/1535-7163.MCT-12-0967. [DOI] [PubMed] [Google Scholar]
- Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol. 2013;3:154. doi: 10.3389/fonc.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saggar JK, Tannock IF. Chemotherapy rescues hypoxic tumor cells and induces their reoxygenation and repopulation - an effect that is inhibited by the hypoxia-activated pro-drug TH-302. Clin Cancer Res. 2015;21:2107–14. doi: 10.1158/1078-0432.CCR-14-2298. [DOI] [PubMed] [Google Scholar]
- Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101:937–49. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- Manallack DT. The pK(a) distribution of drugs: application to drug discovery. Perspect Medicin Chem. 2007;1:25–38. [PMC free article] [PubMed] [Google Scholar]
- Larsen AK, Escargueil AE, Skladanowski A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol Ther. 2000;85:217–29. doi: 10.1016/s0163-7258(99)00073-x. [DOI] [PubMed] [Google Scholar]
- Fais S, De Milito A, You H, Qin W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007;67:10627–30. doi: 10.1158/0008-5472.CAN-07-1805. [DOI] [PubMed] [Google Scholar]
- Schempp CM, von Schwarzenberg K, Schreiner L, et al. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther. 2014;13:926–37. doi: 10.1158/1535-7163.MCT-13-0484. [DOI] [PubMed] [Google Scholar]
- Hernandez A, Serrano-Bueno G, Perez-Castineira JR, Serrano A. Intracellular proton pumps as targets in chemotherapy: V-ATPases and cancer. Curr Pharm Des. 2012;18:1383–94. doi: 10.2174/138161212799504821. [DOI] [PubMed] [Google Scholar]
- Sabolic I, Brown D, Verbavatz JM, Kleinman J. H(+)-ATPases of renal cortical and medullary endosomes are differentially sensitive to Sch-28080 and omeprazole. Am J Physiol. 1994;266(6 Pt 2):F868–77. doi: 10.1152/ajprenal.1994.266.6.F868. [DOI] [PubMed] [Google Scholar]
- De Milito A, Marino ML, Fais S. A rationale for the use of proton pump inhibitors as antineoplastic agents. Curr Pharm Des. 2012;18:1395–406. doi: 10.2174/138161212799504911. [DOI] [PubMed] [Google Scholar]
- Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer treatment development. Cancer Chemother Pharmacol. 2015;75:439–47. doi: 10.1007/s00280-014-2637-z. [DOI] [PubMed] [Google Scholar]
- Tan Q, Joshua A, Saggar J, et al. Effects of pantoprazole to enhance activity of docetaxel against human tumor xenografts by inhibiting autophagy. Br J Cancer. 2015;112:832–40. doi: 10.1038/bjc.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Levy JM, Thompson JC, Griesinger AM, et al. Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. Cancer Discov. 2014;4:773–80. doi: 10.1158/2159-8290.CD-14-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:17163–73. doi: 10.1074/jbc.M114.558288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Zeng P, Kang R, et al. S100A8 contributes to drug resistance by promoting autophagy in leukemia cells. PLoS ONE. 2014;9:e97242. doi: 10.1371/journal.pone.0097242. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Patel KJ, Lee C, Tan Q, Tannock IF. Use of the proton pump inhibitor pantoprazole to modify the distribution and activity of doxorubicin: a potential strategy to improve the therapy of solid tumors. Clin Cancer Res. 2013;19:6766–76. doi: 10.1158/1078-0432.CCR-13-0128. [DOI] [PubMed] [Google Scholar]
- Lee CM, Tannock IF. Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. Br J Cancer. 2006;94:863–9. doi: 10.1038/sj.bjc.6603010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani F, Spada M, De Milito A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96:1702–13. doi: 10.1093/jnci/djh305. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Perut F, Fagioli F, et al. Proton pump inhibitor chemosensitization in human osteosarcoma: from the bench to the patients’ bed. J Transl Med. 2013;11:268. doi: 10.1186/1479-5876-11-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunggal JK, Cowan DS, Shaikh H, Tannock IF. Penetration of anticancer drugs through solid tissue: a factor that limits the effectiveness of chemotherapy for solid tumors. Clin Cancer Res. 1999;5:1583–6. [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–60. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Fung AS, Jonkman J, Tannock IF. Quantitative immunohistochemistry for evaluating the distribution of Ki67 and other biomarkers in tumor sections and use of the method to study repopulation in xenografts after treatment with paclitaxel. Neoplasia. 2012;14:324–34. doi: 10.1593/neo.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–84. [PubMed] [Google Scholar]
- Fais S, Venturi G, Gatenby B. Microenvironmental acidosis in carcinogenesis and metastases: new strategies in prevention and therapy. Cancer Metastasis Rev. 2014;33:1095–108. doi: 10.1007/s10555-014-9531-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolini I, Federici C, Raggi C, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009;284:34211–22. doi: 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federici C, Petrucci F, Caimi S, et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE. 2014;9:e88193. doi: 10.1371/journal.pone.0088193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honscheid P, Datta K, Muders MH. Autophagy: detection, regulation and its role in cancer and therapy response. Int J Radiat Biol. 2014;90:628–35. doi: 10.3109/09553002.2014.907932. [DOI] [PubMed] [Google Scholar]
- Marino ML, Pellegrini P, Di Lernia G, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 2012;287:30664–76. doi: 10.1074/jbc.M112.339127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais S. Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism. J Intern Med. 2010;267:515–25. doi: 10.1111/j.1365-2796.2010.02225.x. [DOI] [PubMed] [Google Scholar]
- Azzarito T, Venturi G, Cesolini A, Fais S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett. 2015;356(2 Pt B):697–703. doi: 10.1016/j.canlet.2014.10.017. [DOI] [PubMed] [Google Scholar]
- De Milito A, Iessi E, Logozzi M, et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007;67:5408–17. doi: 10.1158/0008-5472.CAN-06-4095. [DOI] [PubMed] [Google Scholar]
- De Milito A, Canese R, Marino ML, et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer. 2010;127:207–19. doi: 10.1002/ijc.25009. [DOI] [PubMed] [Google Scholar]
- Zhang B, Yang Y, Shi X, et al. Proton pump inhibitor pantoprazole abrogates adriamycin-resistant gastric cancer cell invasiveness via suppression of Akt/GSK-β/β-catenin signaling and epithelial-mesenchymal transition. Cancer Lett. 2(2 Pt B):704–12. doi: 10.1016/j.canlet.2014.10.016. 356. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wang Y, Li SJ. Lansoprazole induces apoptosis of breast cancer cells through inhibition of intracellular proton extrusion. Biochem Biophys Res Commun. 2014;448:424–9. doi: 10.1016/j.bbrc.2014.04.127. [DOI] [PubMed] [Google Scholar]
- Spugnini EP, Baldi A, Buglioni S, et al. Lansoprazole as a rescue agent in chemoresistant tumors: a phase I/II study in companion animals with spontaneously occurring tumors. J Transl Med. 2011;9:221. doi: 10.1186/1479-5876-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spugnini EP, Buglioni S, Carocci F, et al. High dose lansoprazole combined with metronomic chemotherapy: a phase I/II study in companion animals with spontaneously occurring tumors. J Transl Med. 2014;12:225. doi: 10.1186/s12967-014-0225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagerakis S, Bellile E, Peterson LA, et al. Proton pump inhibitors and histamine 2 blockers are associated with improved overall survival in patients with head and neck squamous carcinoma. Cancer Prev Res (Phila) 2014;7:1258–69. doi: 10.1158/1940-6207.CAPR-14-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Huang SL, Zhang XQ, et al. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway in vitro and in vivo. J Cell Biochem. 2012;113:2474–87. doi: 10.1002/jcb.24122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udelnow A, Kreyes A, Ellinger S, et al. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE. 2011;6:e20143. doi: 10.1371/journal.pone.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ML, Fais S, Djavaheri-Mergny M, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1:e87. doi: 10.1038/cddis.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Klotz U. Proton pump inhibitors: an update of their clinical use and pharmacokinetics. Eur J Clin Pharmacol. 2008;64:935–51. doi: 10.1007/s00228-008-0538-y. [DOI] [PubMed] [Google Scholar]
- Shin JM, Kim N. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogastroenterol Motil. 2013;19:25–35. doi: 10.5056/jnm.2013.19.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brana I, Ocana A, Chen EX, et al. A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and tissue penetration of doxorubicin. Invest New Drugs. 2014;32:1269–77. doi: 10.1007/s10637-014-0159-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression of pharmacodynamic biomarkers γH2AX and cleaved caspase-3 or caspase-6 in EMT6 and MCF7 tumors.