

Summary
The mechanistic target of rapamycin (mTOR) complex 2, mTORC2, is a critical downstream effector of PI3K to stimulate AGC kinase members, including Akt, PKC, and SGK. Liu et al reported that the PH domain of Sin1, an essential component of mTORC2, directly binds the PI3K product PtdIns(3,4,5)P3 to promote mTORC2 kinase activation and membrane localization, thereby revealing a mechanistic link between PI3K and mTORC2.
The mechanistic target of rapamycin (mTOR) is a central cell growth modulating kinase in all eukaryotes. mTOR exists in two different complexes, mTORC1 and mTORC2, which are distinguished by unique accessory protein Raptor and Rictor, respectively. In addition, Sin1 is also a component uniquely present in the mTORC2 and is required for mTORC2 complex formation and function (1–3). Compared to the well-defined regulation and function of mTORC1, mTORC2 is less understood. Akt/PKB, a member of the AGC family kinases, is a key signaling hub in the PI3K pathway and regulates multiple cellular processes such as growth, proliferation, metabolism, and survival. Akt is one of the most important substrates of mTORC2, which promotes Akt activation by directly phosphorylating its hydrophobic motif (Ser473), a site required for AKT maximum activation (4). Phosphorylation at Ser473 primes Akt for further phosphorylation at Thr308 in the catalytic domain by phosphoinositide-dependent protein kinase 1 (PDK1), and thus leading to full activation of Akt. mTORC2 also phosphorylates other AGC kinases, including serum- and glucocorticoid-regulated kinase (SGK) (5) and protein kinase C (PKC) (6). It is generally accepted that mTORC2 is activated by growth factors via PI3K signaling, however, the precise molecular mechanism of mTORC2 activation remains elusive thus far.
Accumulating evidence suggests that Sin1 may play a key role in the regulation of mTORC2. It was found that alternatively spliced Sin1 isoforms in the cell defines distinct mTORC2 pools; of these, only two are regulated by insulin (1). This suggests that Sin1 may function as a mediator between growth factor signaling and mTORC2. Sin1 contains a phospholipid-binding pleckstrin homology (PH) domain that may facilitate the association of mTORC2 with membranes (7).
Recently, Wei and colleagues reported that mTORC2 complex is directly regulated by mTORC1-S6K axis through Sin1 phosphorylation (Fig.1) (8). Phosphorylation of Sin1 at Thr86 and Thr398 by either S6K or Akt dissociates Sin1 from mTORC2, thus resulting mTORC2 inhibition. Importantly, the authors also demonstrated a link between Sin1 phosphorylation and a cancer-derived mutation R81T in Sin1, providing clinical significance of Sin1 phosphorylation in cell growth control. R81T mutation attenuates Sin1 Thr86 phosphorylation and sustains mTORC2 activity and Akt Ser473 phosphorylation upon physiological stimulation, thus bypassing Sin1 phosphorylation-mediated negative regulation of mTORC2 activity in response to upstream PI3K activation (Fig.1) (8).
Figure 1.
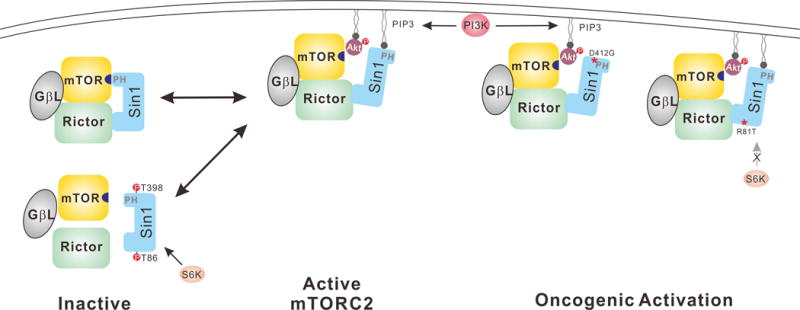
Dual role of Sin1 in mTORC2 regulation. In the absence of stimulation, the PH domain of Sin1 binds to and suppresses mTORC2 activity. Upon PI3K activation, the increased PtdIns(3,4,5)P3 binds to Sin1 PH domain, leading to membrane recruitment as well as relieve of inhibition of mTORC2 by Sin1. The active mTORC2 phosphorylates and activates the membrane associated Akt. Sin1 can be phosphorylated by S6K and Akt to constitutive a negative regulation. Cancer associated mutations in Sin1 can potentiate mTORC1 activity by disrupting the inhibitory phosphorylation in Sin1 or interaction with mTOR.
In the current issue of Cancer Discovery, Liu et al from the same group reported a direct molecular mechanism through which Sin1 regulates mTORC2 kinase activity by PI3K signaling (Fig.1) (9). Although Sin1 is required for mTORC2 integrity and kinase activity, surprisingly the authors found that the PH domain of Sin1 actually binds to mTOR kinase domain and inhibits mTOR activity towards Akt Ser473 phosphorylation. Overexpression of Sin1-PH domain is sufficient to suppress the phosphorylation of Akt Ser473. Interestingly, substitution of Akt-PH with Sin1-PH was able to functionally reconstitute the phosphorylation of Akt Ser473, indicating that the Sin1-PH domain shares biochemical property similar to the Akt PH domain capable of binding PtdIns(3,4,5)P3.
Inspired by the mechanism of PtdIns(3,4,5)P3-mediated Akt activation, the authors indeed demonstrated that PtdIns(3,4,5)P3, a product of PI3K at plasma membrane, is able to interact with Sin1-PH domain (Fig.1). By comparing the Akt1-PH/Ins(1,3,4,5)P4 crystal structure with a computer modeling of Sin1-PH/Ins(1,3,4,5)P4 structure, the authors identified three critical residues including R395, K428 and K464 that mediate PtdIns(3,4,5)P3 binding to Sin1-PH. Mechanistically, increased PtdIns(3,4,5)P3 levels upon insulin or growth stimulation compete with mTOR kinase domain in binding to Sin1-PH, and thus release the Sin-PH inhibition on the mTORC2 activity. Moreover, PtdIns(3,4,5)P3 association with Sin1-PH is also responsible for the recruitment of mTORC2 complex to plasma membrane where mTORC2 can phosphorylate the membrane associated Akt or other physiological substrates.
The significance of this study is further expanded by genetic analysis of cancer-associated mutations in the Sin-PH domain. Analysis of TCGA database revealed several somatic mutations in the Sin1-PH domain. Biochemical characterizations showed that these cancer-associated mutations compromise Sin1-PH binding to mTOR kinase domain, thereby leading to increased mTORC2 activity towards Akt-Ser473 phosphorylation (Fig.1). Among these mutations, D412G shows the most robust enhancement on mTORC2 activity even under non-stimulation conditions, thus identifying a key residue important for interaction with mTOR kinase domain. Interestingly, the cancer mutation analyses show that D412G mutation and R81T mutation, which compromises Sin1 Thr86 phosphorylation (8), are mutually exclusive with either Akt1 gene amplification or PIK3CA oncogenic mutations (9). Collectively, these observations support a notion that mutations in PI3K, Akt, and Sin1 affect a common pathway important for cancer development.
Finally, compared to Sin1-WT, introduction of Sin1-D412G mutant into ovarian cancer cells that is depleted of endogenous Sin1 significantly increased Akt phosphorylation. These cells also show stronger oncogenic features, such as resistance to apoptosis-inducing and chemotherapeutic drugs, enhanced colony formation ability, and increased tumor growth in xenograft. Given that Sin1 phosphorylation is also compromised by a cancer-patient-derived mutation R81T, these findings provide important clues for Sin1 dysregulation in tumorigenesis. Further genetic and biochemical studies are needed to elucidate the mechanisms and relevant contribution of Sin1 mutations in different cancer types.
In summary, Sin1 can regulates mTORC2 in multiple ways: (i) it helps mTORC2 complex formation (1–3); (ii) it is responsible for recruiting certain mTORC2 substrates, including Akt and SGK (10); (iii) its PH domain directly receives the PI3K signal to activate mTORC2 (9); (iv) its PH domain recruits mTORC2 to plasma membrane (9). Notably, Sin1 regulates mTORC2 activity in both positive and negative manners. The switch between negative and positive is dictated by PtdIns(3,4,5)P3. Upon activation of PI3K, binding of the Sin1-PH domain by PtdIns(3,4,5)P3 not only relieves its inhibition on the mTOR kinase but also promotes mTORC2 translocation to plasma membrane for phosphorylation of its physiological substrates. The current study together with previous results establishes Sin1 as a critical signaling integrator for mTORC2 activity.
Acknowledgments
This work is supported by NIH grants GM51586 and CA196878 (K.L.G.).
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current biology: CB. 2006;16:1865–70. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 3.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes & development. 2006;20:2820–32. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) The Biochemical journal. 2008;416:375–85. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 6.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO journal. 2008;27:1919–31. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berchtold D, Walther TC. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Molecular biology of the cell. 2009;20:1565–75. doi: 10.1091/mbc.E08-10-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nature cell biology. 2013;15:1340–50. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-dependent Activation of the mTORC2 Kinase Complex. Cancer discovery. 2015 doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron AJ, Linch MD, Saurin AT, Escribano C, Parker PJ. mTORC2 targets AGC kinases through Sin1-dependent recruitment. The Biochemical journal. 2011;439:287–97. doi: 10.1042/BJ20110678. [DOI] [PubMed] [Google Scholar]