

Abstract
Helicobacter pylori exploits host glycoconjugates to colonize the gastric niche. Infection can persist for decades promoting chronic inflammation, and in a subset of individuals lesions can silently progress to cancer. This study shows that H. pylori chronic infection and gastric tissue inflammation result in a remodeling of the gastric glycophenotype with increased expression of sialyl-Lewis a/x antigens due to transcriptional up-regulation of the B3GNT5, B3GALT5, and FUT3 genes. We observed that H. pylori infected individuals present a marked gastric local proinflammatory signature with significantly higher TNF-α levels and demonstrated that TNF-induced activation of the NF-kappaB pathway results in B3GNT5 transcriptional up-regulation. Furthermore, we show that this gastric glycosylation shift, characterized by increased sialylation patterns, favors SabA-mediated H. pylori attachment to human inflamed gastric mucosa. This study provides novel clinically relevant insights into the regulatory mechanisms underlying H. pylori modulation of host glycosylation machinery, and phenotypic alterations crucial for life-long infection. Moreover, the biosynthetic pathways here identified as responsible for gastric mucosa increased sialylation, in response to H. pylori infection, can be exploited as drug targets for hindering bacteria adhesion and counteract the infection chronicity.
Keywords: Helicobacter pylori, Chronic infection Glycophenotype, Sialic acid-binding adhesin
1. Introduction
Gastric cancer is a public health burden worldwide, being the third leading cause of cancer-related mortality. The Lauren intestinal-type tumors constitute the most frequent form of gastric adenocarcinomas, and commonly arise in the context of a carcinogenic pathway initiated by chronic inflammation of the gastric mucosa triggered by Helicobacter pylori (H. pylori) infection [1]. H. pylori is classified as a carcinogenic agent by the WHO, and epidemiological studies show that 1–3% of infected individuals ultimately develop gastric cancer, which corresponds to a million new cases every year [2].
Disease development depends on bacterial ability to establish close contact with the gastric epithelial cells and to transfer virulence factors [3]. One of the proteins translocated into the host cells is the cytotoxin-associated gene A (CagA) protein, which is phosphorylated by host kinases and interferes with key signal transduction pathways [4]. Individuals infected with cagA-positive strains present increased risk for development of severe disease outcomes, including gastric cancer [5,6].
Bacterial attachment is mediated by outer membrane adhesins that bind to glycoconjugates present in the gastric mucus layer and lining the surface epithelium of the gastric mucosa [7]. The blood group antigen binding adhesin (BabA) recognizes ABO(H)/Lewis b blood group antigens expressed in glycoproteins from the gastro-intestinal tract by secretor individuals [8–10], whereas the sialic acid binding adhesin (SabA) mediates bacterial attachment through binding to α2,3-sialylated structures, such as sialyl-Lewis a (sialyl-Lea) and sialyl-Lewis x (sialyl-Lex) carried by glycosphingolipids and glycoproteins [11]. The babA gene shares regions of high homology with the babB and babC genes. It has been demonstrated that bacteria presenting inactive BabA can gain Lewis b binding properties by babA and babB gene recombination [12]. Recently, a novel H. pylori adhesin has been identified, the LacdiNAc-specific adhesin (LabA), which recognizes di-N-acetyllactosamine (lacdiNAc) motifs, carried by MUC5AC gastric mucin [13]. The myriad of receptors that can be exploited by H. pylori to adhere to the gastric mucosa reflects the multiple target strategy adopted by this bacterium to efficiently colonize the gastric niche and maintain a chronic infection.
Glycan-mediated adhesion of H. pylori to gastric epithelial cells has been shown to act as an important trigger for translocation of bacterial virulence factors into the host cells [14]. The translocation of effector molecules, such as CagA and the bacterial cell wall peptidoglycan (PGN), results in the modulation of different host intracellular signaling pathways, including stimulation of the NF-κB (nuclear factor κB) pathway [15,16].
H. pylori induced inflammatory responses include the up-regulation of proinflammatory cytokines, including IL-8 and TNF-α. Concomitantly with the gastric mucosal inflammation, the human gastric glycosylation patterns change with expression of inflammation associated sialylated glycans [11,17,18]. Importantly, inflammation has been shown to modulate the expression of the glycosyltransferases involved in the biosynthesis of terminal glycan chains [19–21]. However, little is known about the molecular mechanisms governing the glycosylation shift that occurs in gastric mucosa in response to infection.
We have previously shown that H. pylori induces, in human gastric cell lines, the expression of β3GnT5, a GlcNAc-transferase that drives the biosynthesis of the SabA-ligand sialyl-Lex [22]. However, the activation of this glycosylation pathway, as well as the existence of other regulatory mechanisms underlying these glycophenotypic changes, in the complex context of human H. pylori chronic infection, have never been addressed.
To assess the effect of chronic H. pylori infection and gastric mucosa inflammation on the transcriptional regulation of the enzymes that determine the host gastric cell’s glycophenotype, we have evaluated the glycosylation and the glycosyltransferase transcriptomic profile of gastric biopsies from healthy and infected individuals. In addition, we have determined the transcript levels of inflammation markers in gastric tissues and determined the effects of TNF-α proinflammatory cytokine, as well as downstream signaling pathways in the transcriptional regulation of the B3GNT5 gene. Furthermore, we have determined the functional impact of these glycophenotypic alterations on H. pylori ability to attach to the gastric mucosa.
2. Material and methods
2.1. Human gastric tissue samples
This study includes 50 individuals, who were part of a case–control study that encompassed first-degree relatives of patients with early-onset gastric carcinoma (n = 26) and controls that comprised spouses (n = 14) and neighbors (n = 4) of the cases and dyspeptic patients (n = 6) (Supplementary Table 1) [23]. All individuals underwent high definition upper GI endoscopy at Centro Hospitalar do Porto (CHP, Porto, Portugal). Parallel biopsies were collected for RNA extraction, histopathological evaluation and H. pylori culture. Serum samples were also collected from all individuals at the time of endoscopy. This study was approved by the ethical committee of Centro Hospitalar do Porto (Portugal) and written informed consent was received from all participants.
2.2. Histopathological evaluation, H. pylori infection status and cagA genotyping
Gastric biopsy sections were stained with hematoxylin and eosin and modified Giemsa for histological examination by two pathologists who were blinded to the patient data and endoscopic findings. The tissue sections were evaluated based on updated Sydney–Houston classification system. H. pylori was cultured from fresh biopsies and genotyping of cagA was performed as previously described [24]. Individual serological values for anti-H. pylori IgG/IgA were determined according to standard protocol. Cases were classified as H. pylori positive when histology and culture from fresh biopsies were positive. There were two cases classified as H. pylori positive (based on histological observation and high anti-H. pylori IgG/IgA antibody titers) from which bacteria was not possible to cultivate in vitro and therefore the cagA status could not be determined (Fig. 1A and Supplementary Table 1).
Fig. 1.
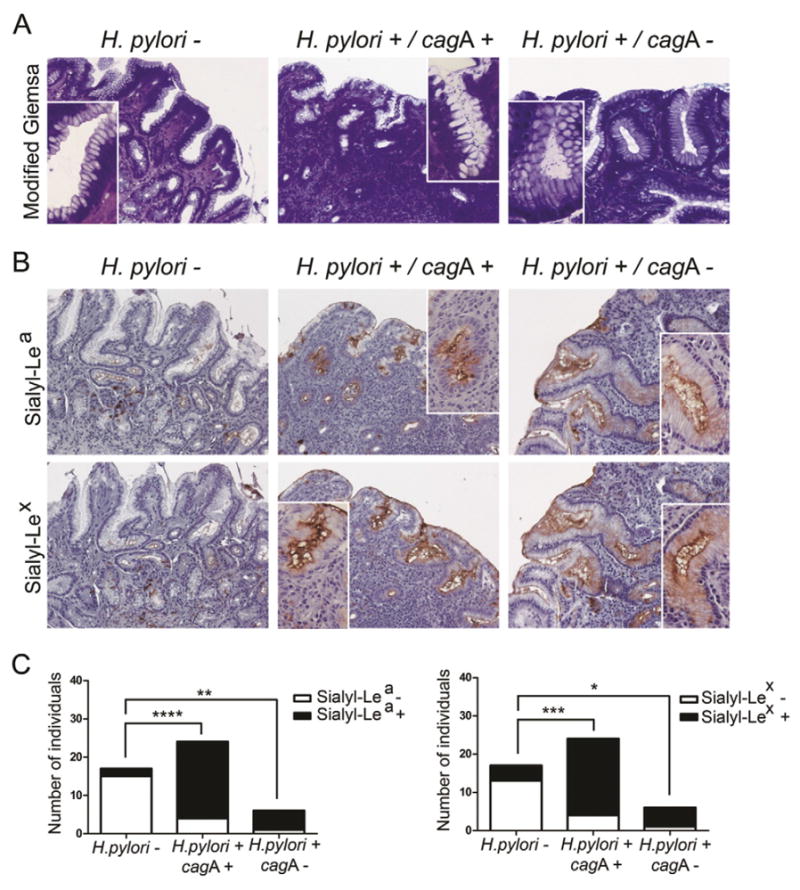
Chronic H. pylori infection of human gastric mucosa results in expression of sialylated antigens. (A) Representative micrographs of H. pylori visualized by modified Giemsa and (B) immunohistochemical detection of sialyl-Lea and sialyl-Lex antigens in paraffin-embedded sections of human gastric mucosa from non-infected individuals (H. pylori−) and individuals infected with either cagA-positive (H. pylori+/cagA+) or cagA-negative (H. pylori+/cagA−) strains. Magnification 200× with inserts of 400×. No major differences were observed regarding the pattern of expression of sialylated antigens in paraffin and frozen sections (data not shown). (C) Graphical representation of sialyl-Lea and sialyl-Lex antigen expression in the three biological groups, statistical significance determined using the Fisher’s exact test *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
2.3. Immunohistochemistry and lectin staining
Tissue staining of sialyl-Lea (CA19.9, Santa Cruz) and sialyl-Lex (KM93, Calbiochem) was performed as previously described [10]. The CA19.9 antibody detects sialyl-Lea and is routinely used in the clinics. Analysis of KM93 glycan epitope specificity showed that it is reactive to sialyl-Lex on core 2, but does not recognizes sialyl-Lex on core 3 [25]. Additionally, binding of KM93 to the non-fucosylated type 2 sialylated structures cannot be excluded as suggested by glycan array analysis from the Consortium for Functional Glycomics (http://www.functionalglycomics.org).
Detection of α2,6-sialylated glycans was performed using the SNA lectin (Sambucus nigra, Vector Laboratories) accordingly to the procedure previously described [10]. Tissue sections from antrum biopsies were used whenever possible. In two cases due to the presence of intestinal metaplasia (IM) in the antrum region, biopsies from incisura angularis were selected. Only cytoplasmic and membrane staining of the foveolar epithelium region was considered for expression evaluation. Expression in secreted mucus and IM areas was excluded from our analysis. Statistical analysis was performed using StatView 5.0 and significance determined using the Fisher’s exact test.
2.4. Gastric biopsy transcriptomic analysis
Quantitative real-time PCR was performed as previously described [26]. Briefly, total RNA was isolated from frozen gastric biopsies using the RNeasy Plus Mini RNA isolation kit (Qiagen). A cDNA reaction for each sample was synthesized according to manufacturer’s instructions using SuperScript III (Invitrogen), including a control reaction lacking reverse transcriptase for detection of contaminating genomic DNA. For qRT-PCR, the cDNA was used in triplicate reactions for each gene tested. The sequences of the primers used in this study are included in Supplementary Table 2. The relative transcript levels in each sample were determined using the ΔΔCt method. Due to the heterogeneous nature of the gastric biopsies, the relative transcript expression of all genes tested was normalized to the expression of the epithelial marker cytokeratin 18 (KRT18). Statistical analysis was performed using GraphPad PRISM 5.0 software and significance was evaluated by unpaired t test with Welch’s correction (95% confidence interval).
2.5. Bioinformatic analysis
Using the Ensembl database [27], the nucleotide sequence corresponding to a CpG island (chr 3: 182970925–182972635) predicted to exist in the 5’UTR region of the human B3GNT5 gene (ENSG00000176597) was extracted and analyzed with the software package PROMO [28] for transcription factor binding site prediction. Default matrices were used and factors predicted within a dissimilarity margin less or equal than 15%, corresponding to NF-κB [T00590] and NF-κB1 [T00593] were selected. The same CpG island sequence was screened for DNase I hypersensitive sites using Ensembl database [27] and nucleotide sequence conservation across the genomes of Homo sapiens, Pan troglodytes, Gorilla gorilla, Pongo abelii, Callithrix jacchus and Mus musculus.
2.6. Human gastric cell line culture
Human gastric carcinoma cell line MKN45, established from a poorly differentiated gastric adenocarcinoma (Japanese Collection of Research Bioresources), was grown in RPMI 1640 medium with Glutamax supplemented with 10% inactive fetal bovine serum and 1% penicillin–streptomycin. This gastric cell line was selected based on previous results from the group showing that this cell line is responsive to H. pylori cagA-mediated effects [22,29] and because it is possible to activate the NF-κB pathway in this cell line by TNF-alpha stimulation.
2.7. TNF-α stimulation and NF-κB pathway inhibition assay
One day prior to treatment, MKN45 gastric cells were seeded under standard conditions into 6-well culture plates (3 × 105 cells/well) and in 8-well chamber slides (1.5 × 104 cells/well) in order to reach 70–80% confluence. Cells were treated with 40 ng/ml TNF-α (Pepro-Tech Inc.) for 20 min, 2 h and 7 h, alone or in combination with 60 μM NF-κB activation inhibitor IV (481412, Calbiochem). As control, the same cell line was cultured only with 60 μM NF-κB activation inhibitor IV or without treatment, for the same time periods. For each condition tested, three biological independent experiments were performed, each with three replicates.
2.8. Immunofluorescence staining
Cells grown on 8-well chamber slides were fixed with 4% parafor-maldehyde for 15 min at room temperature, permeabilized in 0.1% Triton X-100 in PBS for 10 min and incubated with goat non-immune serum diluted 1:5 in PBS with 10% of BSA. Cells were then incubated during 2 h at room temperature with NF-κB p65 Rabbit mAb primary antibody (D14E12 Cell Signaling) diluted 1:150 in PBS containing 5% of BSA. Following washing with PBS, cells were incubated with Alexa Fluor 488 goat anti-rabbit immunoglobulin (Invitrogen), diluted 1:500 in PBS with 5% of BSA for 1 h at room temperature. Cells were washed in PBS, incubated with DAPI (Sigma) for 10 min and mounted in VectaShield (Vector Laboratories). Images were acquired using a Zeiss Axio cam MRm and the AxioVision Rel. 4.8 software.
2.9. B3GNT5 qRT-PCR
Total RNA was extracted from cell lines using TRI reagent LS (Sigma-Aldrich), according to manufacturer’s protocol. RNA yield and quality were determined spectophotometrically and 5.0 μg of total RNA was reverse transcribed using Superscript III (Invitrogen), as described above. Expression of B3GNT5 was quantified using Taqman probes, acquired as pre-developed assays from Applied Biosystems (HS00908059_m1) and normalized to the expression of the endogenous control 18S (HS99999901_s1). Each sample was amplified in triplicate in an ABI Prism 7500 (Applied Biosystems). Relative transcript levels were determined using the ΔΔCT-method. Statistical analysis was performed using GraphPad PRISM 5.0 software and significance was evaluated with Student’s T-test.
2.10. H. pylori culture
The H. pylori strains 17875/Leb and 17875babA1∷kan babA2∷cam (17875babA1A2) were grown in Pylori agar (BioMérieux, Marcy l’Étoile, France) at 37 °C under microaerobic conditions. For strain 17875babA1∷kan babA2∷cam, culture media included also 20 mg/L Chloramphenicol (Sigma) and 25 mg/L Kanamycin (Sigma). The 17875/Leb strain is a spontaneous mutant that binds Leb but does not bind to sialylated antigens [11]. Clinical isolates from patients were cultured as previously described [30].
2.11. Immunoblot for BabA, BabB and SabA outer membrane proteins
Bacteria were collected from plates using 1 mL PBS and centrifuged at 2400 g for 5 min; proteins were extracted with the lysis buffer RIPA containing 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor cocktail. Protein concentration was determined using the BCA protein assay kit and 50 μg was loaded in acrylamide gel (stacking 5%/resolving 10%) for electrophoresis. Proteins were transferred to immunoblot polyvinylidene difluoride membranes (Hybond-P PVDF Membrane, Amersham Biosciences) at 50 V for 1 h. Membranes were blocked for 1 h in PBS-Tween containing 5% nonfat dried milk prior to incubation overnight at 4 °C with either BabA (AK277 1:10,000), BabB (AK276 1:3000) or SabA (AK278 1:5000) antibodies [31]. Blots were incubated with a HRP-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology) and finally developed with ECL (Amersham ECL Western Blotting detection reagents).
2.12. H. pylori binding assay to human gastric mucosa
Labeling with FITC and adhesion assays were performed as previously described [8,10,11]. Evaluation of bacterial binding was estimated by the number of adhered bacteria to superficial foveolar epithelium region under 200× magnification. Three independent gastric biopsies tissue samples were considered for each biological group and at least 24 different fields from each section were quantified using the ImageJ software. Statistical analysis was performed using GraphPad PRISM 5.0 software and significance was evaluated by one-way ANOVA with Dunnett’s Multiple Comparison Test (95% confidence interval).
3. Results
3.1. H. pylori chronic infection induces the expression of alpha2,3-sialylated antigens in human gastric mucosa
The expression of the type 1 and type 2 α2,3-sialylated antigens, sialyl-Lea and sialyl-Lex respectively, was evaluated by tissue immunolabeling in a series of 50 gastric biopsies that have been characterized regarding gastric tissue histopathological features and genetic characteristics of H. pylori infecting strains (Supplementary Table 1).
As represented in Fig. 1, the majority of the non-infected gastric mucosa showed no expression of sialylated-Lewis antigens, whereas most of the H. pylori infected individuals presented both sialyl-Lea (85%) and sialyl-Lex (85%) staining, demonstrating that the expression of these α2,3-sialylated structures is significantly associated with H. pylori infection status (p-value < 0.0001) (Fig. 1B, C, Supplementary Table 3). The pattern of staining with CA19.9 (anti-sialyl-Lea mAb) and KM93 (anti-sialyl-Lex mAb) was similar. Both antibodies showed immunoreactivity in surface mucous cells as well as in few cells from the antrum glands. Though, the extent and intensity of sialyl-Lex staining was higher, in comparison with sialyl-Lea, with strong apical labeling of most surface epithelial cells lining the gastric mucosa (Fig. 1B). No differences were observed regarding the pattern of expression of sialylated antigens in paraffin and frozen sections (data not shown).
Our results suggest that the expression of α2,3-sialylated-Lewis antigens is independent of the cagA status of the infecting strain, since individuals infected with either cagA-positive or cagA-negative strains presented significantly higher levels of both sialyl-Lea and sialyl-Lex than the non-infected samples (Fig. 1B, C and Supplementary Table 3).
In addition, the expression of α2,6-sialylated antigens was evaluated using the SNA lectin in a subseries of 15 biopsies including 5 controls and 10 H. pylori infected individuals (5 cagA+ and 5 cagA −). No strong binding of α2,6-specific SNA lectin was observed in gastric tissue from both controls and infected individuals (Supplementary Fig. 1). A weak positive staining was observed in rare epithelial cells of 3 H. pylori negative and 1 H. pylori infected samples. As expected, SNA positivity was observed in inflammatory cells of the gastric mucosa (Supplementary Fig. 1).
3.2. Transcriptional up-regulation of glycosyltransferase genes by H. pylori chronic infection
To determine how H. pylori chronic infection and mucosal inflammation regulate the human enzymatic pathways that define the gastric glycophenotype, we have evaluated the transcript abundance of 19 glycosyltransferases involved in glycan biosynthesis (Fig. 2A), in gastric biopsies from healthy and H. pylori infected individuals. This analysis included a subseries of 24 individuals (H. pylori negative n = 8 and H. pylori positive n = 16) from which frozen gastric biopsies were available (Supplementary Table 1). The transcriptional analysis was designed to take into consideration the representation of epithelial cells in the biopsies samples by normalization using the epithelial marker cytokeratin 18 (KRT18).
Fig. 2.
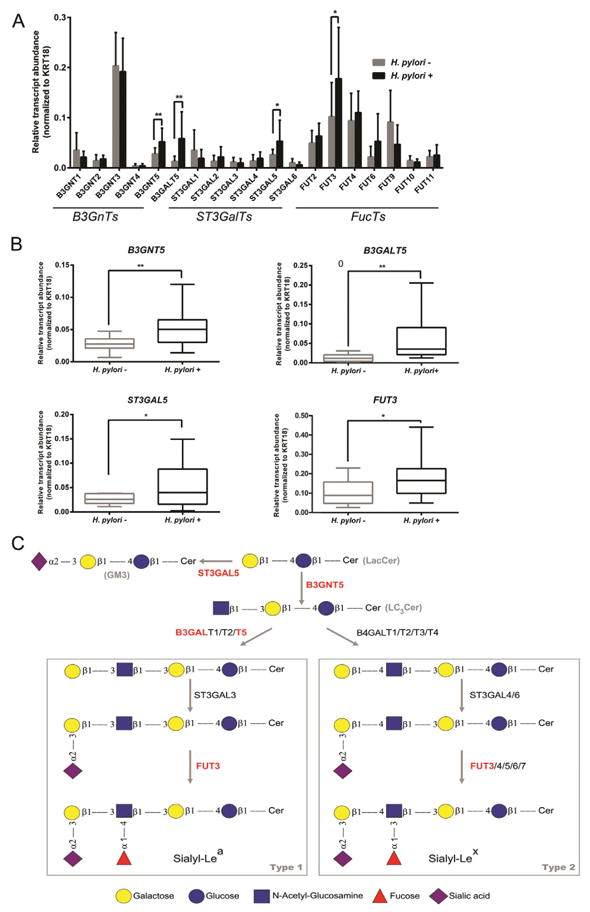
Transcriptomic analysis of glycosyltransferase genes involved in biosynthesis of glycan chains in human gastric mucosa. (A) qRT-PCR analysis of glycosyltransferases, including GalNAc-transferases, Gal-transferases, Sialyl-transferases and Fucosyl-transferases in human gastric biopsies. Each bar represents the mean value of relative transcript abundance considering all individuals within each biological group and error bars represent the standard deviation. (B) qRT-PCR analysis of B3GNT5, B3GALT5, ST3GAL5 and FUT3 genes. The box and whisker plots represent relative gene transcript abundance for each biological group including the minimum and maximum values and the median. qRT-PCR reactions were performed in triplicate and relative transcript abundance of the target gene was normalized to KRT18 expression levels. Statistical significance determined using unpaired t-test with Welch correction.*p < 0.05 and **p < 0.001. (C) Cartoon representation of the pathways involved in the biosynthesis of sialylated-Lewis type 1/2 chains in glycosphingolipids. The genes represented in these pathways that are shown to be induced in response to H. pylori infection are represented in red. Some of the enzymes depicted in the scheme involved in terminal sialylation and fucosylation may also use as acceptors type 1/2 chains on glycoproteins (not represented in this cartoon).
The evaluation of the expression of the β1,3-N-acetylglucosaminyltransferase (B3GnTs) family of genes showed that B3GNT5 transcript expression was significantly increased in H. pylori infected individuals (p < 0.001), whereas the relative transcript levels of B3GNT1, 2, 3 and 4 were similar in the two biological groups (Fig. 2A, B).
In addition, the transcriptomic analysis of gastric biopsies showed that H. pylori infected individuals presented significantly higher transcript levels of the β1,3-galactosyltransferase 5 gene (B3GALT5) (p < 0.001), that encodes a Gal-transferase involved in the addition of a galactose residue to nascent type 1 chains (Fig. 2A, B and C).
The terminal modification of Lewis antigens structures is mediated by sialyl- and fucosyltransferases (Fig. 2C). We have analyzed the expression of all members of the β-galactoside α2,3-sialyltransferase family (ST3GalTs), and only ST3GAL5 gene showed different expression in the two biological groups, with H. pylori infected individuals presenting significantly higher transcript levels than the controls (p < 0.05) (Fig. 2A, B).
Regarding the α1,2-(FUT2) and α1,3/4-(FUT3, FUT4, FUT6, FUT9, FUT10 and FUT11) fucosyltransferase families, responsible for terminal fucosylation, our analysis showed that only FUT3 expression was significantly increased in H. pylori infected individuals (p < 0.05) (Fig. 2A, B).
3.3. Gastric mucosal local transcript levels of proinflammatory cytokines
To assess the inflammatory status of the gastric mucosa samples, we evaluated the transcript levels of proinflammatory markers described to be induced by H. pylori infection. As expected, we observed that, despite inter-individual variation, H. pylori infected individuals present a markedly enhanced inflammatory profile, with significantly higher transcript levels of IL-6, IL-8, TNF-α and IFN-μ molecules (Fig. 3). Importantly, and in agreement with the histology data on inflammation and infiltration of polymorphonuclear cells, individuals in the non-infected group presented barely detectable levels of these proinflammatory molecules (Fig. 3 and Supplementary Table 1). No significant differences were observed for IL-1β transcript levels in the two biological groups (Fig. 3).
Fig. 3.
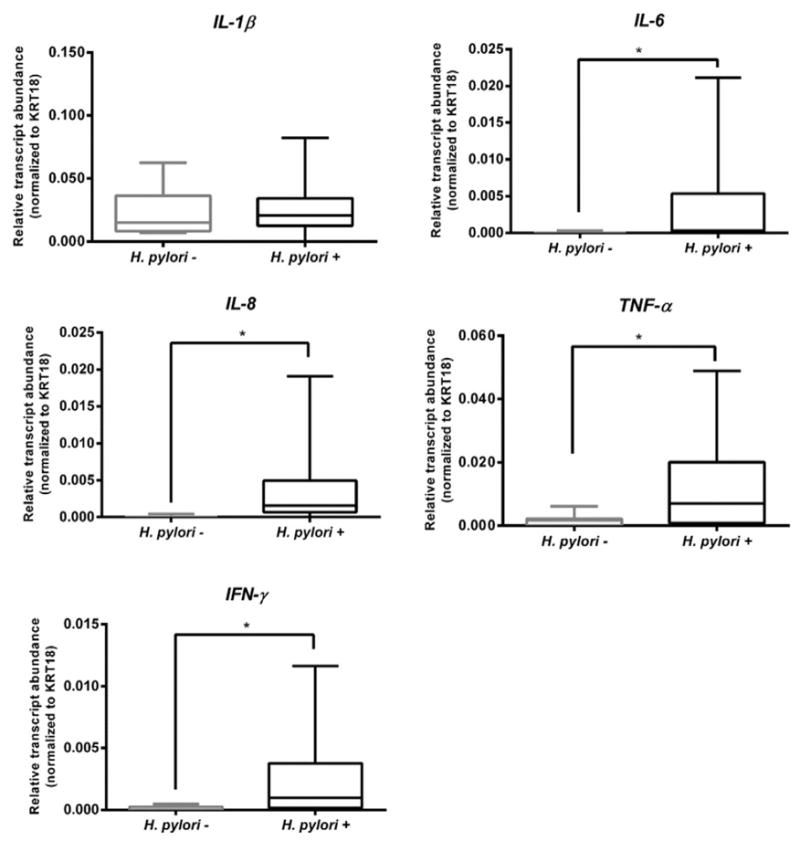
Gastric mucosal local transcript levels of inflammatory markers. qRT-PCR analysis of IL-1β, IL-8, IL-6, TNF-α and IFN-γ genes. The box and whisker plots represent relative gene transcript abundance for each biological group including the minimum and maximum values and the median. qRT-PCR reactions were performed in triplicate and relative transcript abundance of the target gene was normalized to KRT18 expression levels. Statistical significance determined using unpaired t-test with Welch correction.*p < 0.05.
3.4. Effect of TNF-α induced activation of NF-κB pathway on transcriptional regulation of B3GNT5
The increased levels of TNF-α detected on gastric mucosa from H. pylori infected individuals (Fig. 3) were consistent with our previous data demonstrating that exposure of gastric epithelial cells to this proinflammatory cytokine led to increase expression of B3GNT5 [22]. We further investigated the involvement of NF-κB pathway on the expression of B3GNT5, as a downstream response to TNF-α activation.
In silico analysis showed that a CpG island is predicted to exist in the 5’ start of the B3GNT5 gene (chr 3: 182970925–182972635, Fig. 4A, Ensembl v75 [27]). This sequence, putatively encompassing the B3GNT5 promoter region [32], was shown to include several putative binding sites for the transcription factors NF-κB and NF-κB1 (2 and 4 predicted binding sites, Fig. 4A1, 2 [28]). Moreover, such binding sites were predicted to correspond to open chromatin areas, as pointed out by the presence of overlapping DNase-I hypersensitive sites [27], suggesting availability for transcription factor binding (Fig. 4A2). Additionally, most of the putative binding sites identified corresponded to sequences highly conserved between the genomes of human and five primates, an important indicator of biological functional relevance (Fig. 4A5). Comparative analysis of human and mouse genomes indicated that sequence conservation was reduced, suggesting a potential for differential regulation in distant species.
Fig. 4.
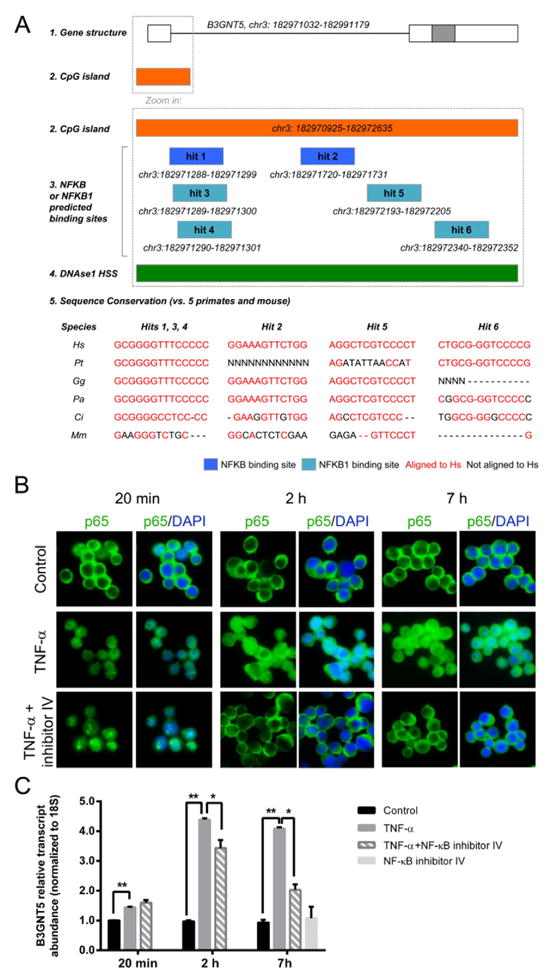
Regulation of B3GNT5 gene expression. (A) Schematic representation of B3GNT5 gene structure (A1: white boxes for untranslated regions, gray box for protein-coding regions [27]) and predicted CpG island (A2 [27]), showing 6 putative NF-κB (in dark blue) or NF-κB1 binding sites (in light blue, A3), all of which overlapping with open chromatin areas according to DNAse I hypersensitivity sites (in green, A4 [27]). Also displayed are the results for the NF-κB or NF-κB1 putative binding sites sequence conservation analysis across human, 5 primates and mouse: in red, conserved nucleotides; in black, nucleotides or gaps not conserved with the human sequence (Hs for Homo sapiens, Pt for Pan troglodytes, Gg for Gorilla gorilla, Pa for Pongo abelii, Cj for Callithrix jacchus and Mm for Mus musculus, A5). (B) Immunofluorescence labeling for p65 (NF-κB-pathway element) in MKN45 cell line stimulated with TNF-α in the absence or presence of NF-κB activation inhibitor IV at 20 min, 2 h or 7 h time points. Cell nuclei are stained with DAPI. Magnification 630×. (C) qRT-PCR analysis of B3GNT5 gene expression in MKN45 cell line stimulated with TNF-α in the absence or presence of NF-κB activation inhibitor IV at 20 min, 2 h or 7 h time points. The graph represents average value and SD of two independent experiments performed in triplicate. Significance was evaluated with Student’s T-test. *p < 0.05 and **p < 0.001.
TNF-α treatment of MKN45 gastric cells resulted in nuclear translocation of the p65 NF-κB subunit (Fig. 4B). The intensity of p65 nuclear staining was higher at 2 and 7 h after TNF-α addition to the cells culture medium and at these two time-points a 4-fold increase in B3GNT5 gene expression was observed (Fig. 4C).
Concomitant treatment of MKN45 cells with TNF-α and NF-κB activation inhibitor IV (481412) efficiently inhibited p65 nuclear translocation at the 2 and 7 h time-points (Fig. 4B). Furthermore, evaluation of B3GNT5 transcript levels showed that NF-κB activation inhibitor IV led to a significant impairment of TNF-α-mediated up-regulation of B3GNT5 transcription, and this effect was more pronounced at 7 h post treatment with NF-κB activation inhibitor IV (Fig. 4C). The p65 nuclear staining observed at the 20 min time-point in the presence of the inhibitor, reflects the lack of the required time for suppression of NF-kappaB signaling and as expected at this time point B3GNT5 transcription levels were not reduced. MKN45 cells treated only with NF-κB activation inhibitor IV were similar to control cells in all assays.
3.5. Gastric mucosa sialylation controls SabA-mediated H. pylori adhesion
Immunoblot analysis of H. pylori clinical isolates from chronically infected subjects showed that most strains presented the expression of both BabA and BabB proteins (Fig. 5A). Expression of the sialic acid binding adhesin, SabA, was more heterogeneous among the clinically isolated strains, with 13 out 25 strains showing very low levels or absence of SabA protein expression. For some clinical isolates a double band was observed, this may stem from variations in protein size. No association was observed between SabA expression, evaluated by immunoblot analysis of clinical isolates in in vitro cultures, and sialyl-Lea/x expression in the gastric tissue from the corresponding subject (Fig. 5A).
Fig. 5.
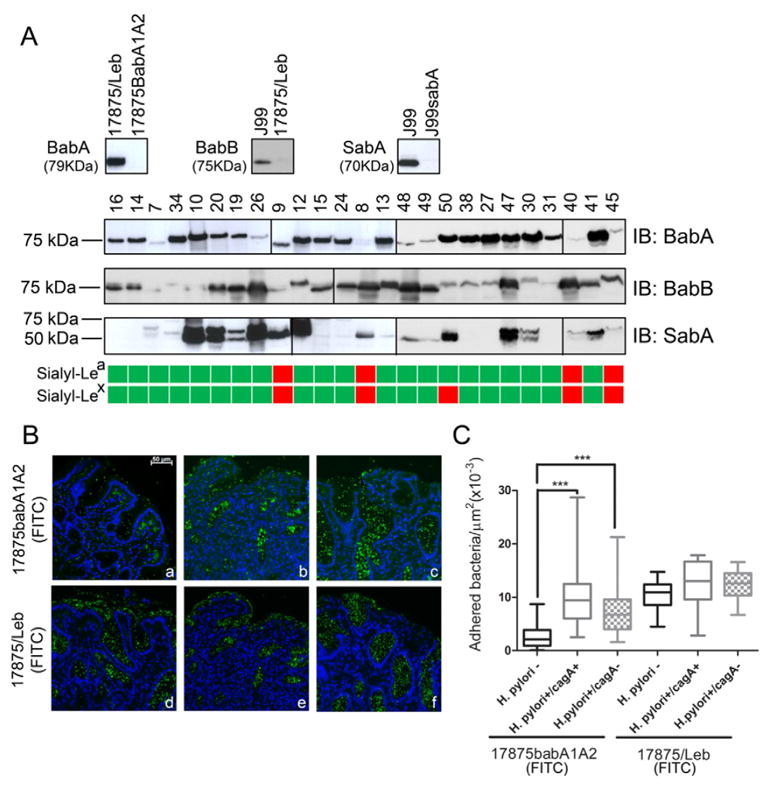
Impact of increased sialylation on H. pylori attachment to inflamed human gastric mucosa. (A) Immunoblot analysis of model H. pylori strains and clinical isolates using BabA, BabB and SabA recognizing antibodies, and schematic representation of sialyl-Lea and sialyl-Lex expression in the corresponding individuals gastric biopsies (green: positive; red: negative). (B) Adhesion of fluorescein-labeled H. pylori strain 17875babA1A2 (functional SabA+) and 17875/Leb (functional SabA −) to gastric mucosa tissue sections from non-infected (H. pylori−) (a, d), H. pylori CagA(+) strains infected (H. pylori+/CagA+) (b, e) and H. pylori CagA(−) strains infected (H. pylori+/CagA−) (c, f) individuals, magnification 200×. (C) Quantification of bacterial adhesion to human gastric mucosa tissue samples, the box and whisker plots represent the minimum and maximum values and the median of at least 24 different fields from three independent gastric biopsies for each biological group; significance was determined by one-way ANOVA with Dunnett’s Multiple Comparison Test. ***p < 0.001.
In order to understand the impact of the altered glycophenotype induced upon H. pylori infection on bacterial attachment to inflamed gastric mucosa, we further evaluated the adhesion of fluorescein-labeled bacteria to gastric biopsies. We have used two previously described model strains, the 17875babA1A2 mutant which lacks BabA and therefore adherence is mediated by the SabA adhesin, and the 17875/Leb spontaneous mutant strain that expresses a functional BabA adhesin but it is unable to bind sialylated structures [11]. As shown in Fig. 5 (B and C), the 17875babA1A2 strain (SabA competent) adhered significantly more to the surface mucous cells and glands of gastric mucosa from H. pylori infected individuals displaying sialylated antigen expression (b, c), than to tissue sections from non-infected subjects without sialylated antigens expression (a) (p < 0.001). In contrast, the strain 17875/Leb (BabA competent), adhered to a similar extent to gastric mucosa from non-infected and H. pylori infected individuals (d, e, f), independent of the sialylation patterns of the gastric mucosa (Fig. 5B, C). Despite lack of association between SabA expression and mucosal expression of sialylated antigens, we observe that SabA-mediated binding is significantly associated with sialylated antigens expression. These results are in agreement with previous data showing that SabA binding is associated with sialyl-Lex expression levels and several markers of tissue inflammation [11].
4. Discussion
In the present study, we demonstrate that H. pylori chronic infection results in a remodeling of the gastric mucosa glycosylation profile, with de novo expression of the α2,3-sialylated structures, sialyl-Lea and sialyl-Lex. In agreement with our data, the increased expression of α2,3-sialylated structures in response to H. pylori has been described to occur in humans [11,17] and in different animal models of infection [11,33–35]. Noteworthy, no alteration in α2,6-sialylated glycans [36] was observed, when comparing infected individuals and controls, supporting that H. pylori specifically promotes α2,3-sialylated antigens biosynthesis.
The biosynthesis of terminal sialylated Lewis structures is dependent on the coordinated activity of several glycosyltransferases. We have previously shown, using in vitro gastric cell line models, that H. pylori is able to induce B3GNT5 expression [22]. This observation is now validated in the context of human H. pylori chronic infection by the demonstration that gastric biopsies from H. pylori infected individuals present significantly higher transcript levels of B3GNT5 (Fig. 2A, B). The B3GNT5 gene encodes a glycosyltransferase responsible for transfer a N-acetylglucosamine (GlcNAc) to galactose (Gal), leading to the biosynthesis of lactotriaosylceramide (Lc3Cer: GlcNAc(β1,3)Gal(β1,4)Glc-ceramide), the precursor structure of lacto- (type 1) and neolacto-series (type 2) carbohydrate chains on glycosphingolipids (Fig. 2C) [37,38]. Importantly, B3gnt5-deficient mice, completely lack Lc3Cer synthase activity, demonstrating that β3GnT5 expression is required for lacto/neolacto-series glycosphingolipids biosynthesis [39]. It was also shown that sialyl-Lex biosynthesis in human colon cancer cells is determined by Lc3Cer precursor backbone chain biosynthesis [40,41]. In agreement, we have demonstrated in vitro that overexpression of β3GnT5 is sufficient to increase sialyl-Lex expression [22]. Interestingly, in vitro we observed that β3GnT5 induction was associated with the H. pylori cagA status, however in the mucosal samples such association was not observed, indicating that within the gastric microenvironment, B3GNT5 transcription is not strictly dependent on CagA injection and that in the context of chronic infection B3GNT5 expression can be induced by the host inflammatory response. This observation further suggests that in vitro models of infection due to lack of inflammatory context may have limitations and supports the importance of validation using clinical samples.
In addition H. pylori infected subjects showed an up-regulation of the B3GALT5 gene (Fig. 2A, B). Enhanced expression of β1,3-galactosyltransferase 5 (β3GalT5) has been reported to result in increased biosynthesis of extended type 1 chains on lactosylceramides and glycoproteins in colon carcinoma cells [42,43], while suppression of β3GalT5 in pancreas adenocarcinoma cells reduced the expression of sialyl-Lea [44]. In line with these observations, the induction of β3GalT5, in response to H. pylori infection, can drive the extension of precursor chains towards the biosynthesis of type 1 sequences, therefore contributing for the increased expression of sialyl-Lea (Fig. 2C).
The transfer of sialic acid to terminal Gal on Lewis structures is mediated by α2,3-sialyltransferases. The ST3Gal III preferentially uses type 1 chains as acceptors leading to sialyl-Lea biosynthesis, whereas ST3Gal IV and ST3Gal VI modify preferentially type 2 sequences to produce sialyl-Lex [45]. No differences were observed regarding the transcript levels of the genes encoding these enzymes in gastric biopsies, and in H. pylori infected gastric cell lines [22], supporting the hypothesis that it is the accumulation of type 1 and 2 lacto-series precursor chains, rather than increased sialylation that leads to increased expression of terminal sialylated Lewis structures in response to infection. Noteworthy, we observed that ST3GAL5 transcription was up-regulated in H. pylori infected individuals (Fig. 2A, B). This gene encodes a sialyltransferase, ST3Gal V, described to use almost exclusively lactosylceramide (LacCer) as acceptor, leading to GM3 ganglioside biosynthesis (Fig. 2C) [45]. Although GM3 ganglioside is not involved in H. pylori binding [46,47], it was demonstrated that GM3 has a good neutralizing capacity against the H. pylori vacuolating toxin VacA, preventing its entrance into the gastric cells [48]. Hence, the induction of ST3GAL5 may reflect a host response to decrease the epithelial damage associated with the infection.
Our data shows that H. pylori infected individuals also present significantly higher transcript levels of FUT3 (Fig. 2A, B), whereas no alterations in transcript levels were observed in any of the other fucosyltransferases. The increased expression of FUT3 is biologically relevant since α1,3/4-fucosylation of sialyl-lactose and sialyl-lactosamine is known to lead to the biosynthesis of sialyl-Lea/x (Fig. 2C) and these structures have been demonstrated to be better receptors for H. pylori than its precursor non-fucosylated glycan chains [49]. The up-regulation of sialyl-Lex expression in the gastrointestinal tissue, particularly in colon, may also be attributed to decreased expression of the B4GALNT2 glycosyltransferase, responsible for Sda antigen biosynthesis [50,51]. Further evaluation of the relevance of this molecular mechanism during gastric carcinogenesis remains to be fully addressed [51].
There is a large collection of data supporting that modulation of host glycosylation profile is associated with infection and host inflammatory response [52]. Chronic gastric inflammation induced by H. pylori is characterized by the up-regulation of diverse cytokines and chemokines [53].
The transcriptomic analysis showed that H. pylori infected individuals present a local enrichment in the proinflammatory cytokines IL-6, IL-8, TNF-α and IFN-γ (Fig. 3), that was accompanied by the recruitment of inflammatory cells to the inflamed tissue (Supplementary Table 1). We did not observe a significant up-regulation of IL-1β transcript levels (Fig. 3), in contrast with the moderate increase previously reported in gastric mucosal samples of H. pylori infected individuals [54,55].
Based on our previous observations that TNF-α stimulation of gastric epithelial cells resulted in increased B3GNT5 expression [22], we have investigated the molecular mechanism underlying TNF-mediated induction of B3GNT5. TNF-α is recognized as a potent activator of the canonical NF-κB pathway [56]. Moreover, according to our bioinformatic analysis, the predicted B3GNT5 promoter sequence presents several highly conserved putative binding sites for NF-κB transcription factors in open chromatin areas (Fig. 4A). Under resting conditions NF-κB complexes are known to be sequestered in the cytoplasm by inhibitory IκB proteins [56]. Upon stimulation, namely by tumor necrosis factor receptors family signaling upon TNF-α binding, IκB is phosphorylated and targeted for proteosomal degradation, releasing p65-containing heterodimers to translocate to the nucleus and stimulate the transcription of specific genes [56]. Several reports have described activation of the NF-κB pathway in response to H. pylori infection and inflammation [57]. Additionally, we have previously shown that H. pylori infection results in induction of several genes downstream of the NF-κB pathway in gastric cells [22]. We demonstrated that inhibition of the NF-κB pathway, assessed by the impairment of p65 nuclear translocation, antagonized the effect of TNF-α on activation of B3GNT5 transcription (Fig. 4B, C). In line with our observations, a global gene expression analysis showed that blocking of NF-κB-mediated responses in endothelial cells led to the down-regulation of B3GNT5 [58]. Altogether, these data indicate that the increased expression of B3GNT5 induced by TNF-α is mediated through activation of the NF-κB pathway (Fig. 6).
Fig. 6.
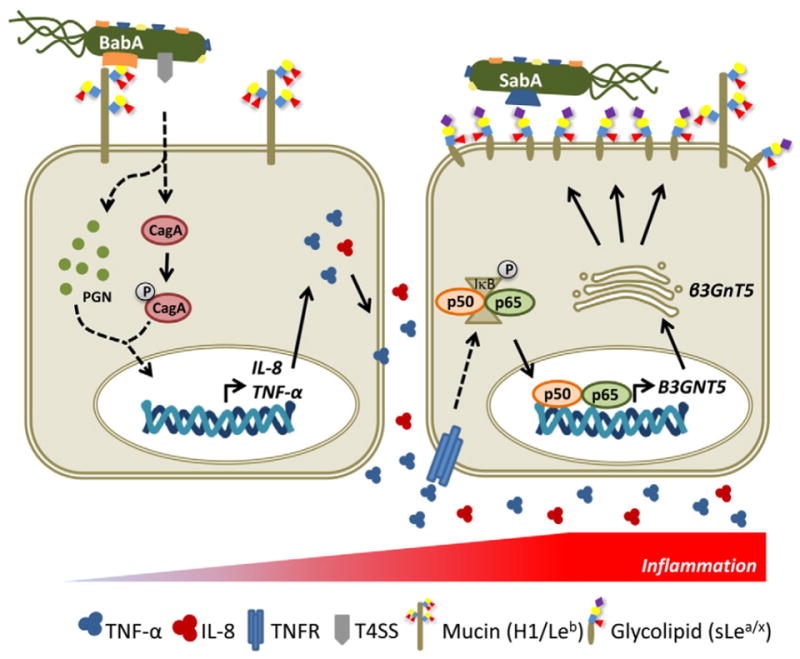
A model for modulation of the gastric mucosal glycophenotype during Helicobacter pylori chronic infection and implications in bacterial adhesion and pathogenesis. In healthy gastric mucosa, H. pylori adhesion to the epithelial cells is mainly mediated by BabA recognition of H-type 1 (H1) and Lewis b (Leb) structures present on glycoproteins (mainly mucins) expressed on gastric surface epithelium. This glycan-mediated adhesion favors translocation of bacterial virulence factors, such as CagA and peptidoglycan (PGN), through the type IV secretion system (T4SS) into the host cells, leading to the activation of several intracellular signaling pathways that culminate in activation of transcription of proinflammatory cytokines, including IL-8 and TNF-α. We propose that during chronic infection and gastric mucosa inflammation, the increased levels of TNF-α result in stimulation of the NF-κB canonical pathway, with translocation of p65 to the nucleus and activation of B3GNT5 gene transcription. Although not depicted in this illustration, it cannot be excluded direct activation of NF-κB pathway by bacterial products (CagA, PGN) or activation by TNF-α secreted by recruited inflammatory cells. The up-regulation of β3Gnt5 activity leads to increased biosynthesis of terminal sialylated type 1/2 structures in glycosphingolipids, resulting in a remodeling of the gastric epithelial cells glycophenotype. The increased expression of the inflammation-associated sialylated structures, recognized by the sialic acid binding adhesin (SabA), promotes a closer membrane attachment of H. pylori to inflamed gastric mucosa, an important feature for infection chronicity.
Long-term colonization of the gastric mucosa and the establishment of a chronic infection are dependent on balanced adaptation of both host and H. pylori [59]. H. pylori exploits multiple glycan targets to efficiently colonize the gastric mucosa, taking benefit of a large collection of outer membrane proteins presenting lectin activity, namely the BabA, LabA and SabA adhesins. Moreover, H. pylori adherence features are known to be dynamic, allowing the bacteria to adapt to gastric microenvironment changes, such as altered glycosylation profiles [11]. In our analysis, 48% of the clinical H. pylori isolates present SabA protein expression (Fig. 5A), which is in accordance with the SabA expression frequency described on European populations [11,31,49]. Importantly, SabA expression has been demonstrated to be subject to a dynamic on–off switching regulated by different genetic mechanisms, including slipped-strand mispairing (SSM) [11], a repetitive DNA element regulation by a rheostat-like mechanism [60], gene duplication by gene conversion [61], and by the acid-responsive signal (ArsRS) two component signal transduction regulatory system [62]. This rapid switch of SabA expression may explain the lack of association between SabA protein expression and sialyl-Lea and sialyl-Lex tissue levels (Fig. 5A). While the individual gastric sialylation glycome is maintained once a chronic infection is established, the SabA expression is expected to differ within an individual over time.
We have further evaluated the impact of increased sialylation, observed in H. pylori chronically infected patients, on bacterial attachment to gastric mucosa. Our results demonstrated that adhesion of 17875babA1A2 strain, expressing an active SabA adhesin, was significantly higher in gastric tissue samples displaying higher sialyl-Lea/x expression (Fig. 5B, C). This higher binding potential can be attributed to SabA-mediated binding to sialylated receptors since adhesion of a strain lacking a functional SabA adhesin, 17875/Leb strain, showed no differences in adhesion. SabA-mediated adhesion to inflamed gastric mucosa is particularly relevant, since this adhesin binds sialylated antigens mainly present on membrane glycosphingolipids [11], therefore promoting a more intimate contact between the bacteria and the host cells for a tighter fit of the infection load and efficient transfer of bacterial virulence factors (Fig. 6).
5. Conclusions
In summary, we demonstrate, for the first time, in human gastric biopsies that H. pylori exploits the tightly regulated host cell glycosyla-tion machinery inducing the expression of B3GNT5, B3GALT5 and FUT3 and leading to biosynthesis of SabA-ligands. Furthermore, we show that up-regulation of B3GNT5, which encodes a key enzyme in inflammation-driven gastric glycophenotype modulation, results from TNF-α-mediated activation of the NF-κB pathway. Finally, our results demonstrate that modulation of the host gastric glycosylation profile favors H. pylori chronic infection by contributing to bacterial epithelial attachment to inflamed gastric mucosa.
Supplementary Material
Acknowledgments
We thank Prof. Thomas Borén and Dr. Jeanna Bugaytsova for critically discussing the manuscript. We thank Prof. Carlos Lopes for histopath-ological analysis.
Funding
IPATIMUP integrates the i3S Research Unit, which is partially supported by FCT, the Portuguese Foundation for Science and Technology (PEst C/SAU/LA0003/2013). This work is funded by FEDER funds through the Operational Programme for Competitiveness Factors-COMPETE (NORTE 07 0124 FEDER 000024; FCOMP-01-0124-FEDER 028188; FCOMP-01-0124-FEDER 041276) and National Funds through the FCT-Foundation for Science and Technology (EXPL/CTM-BIO/ 0762/2013, PTDC/BBB-EBI/0786/2012) and acknowledges support by the European Union (Seventh Framework Programme GastricGlycoExplorer project, grant number 316929). Grants were received from FCT, POPH (Programa Operacional Potencial Humano) and FSE (Fundo Social Europeu) (SFRH/BPD/75871/2011 to AM; SFRH/SINTD/60034/2009 to RMP; SFRH/BPD/84084/2012 to RMF; SFRH/BPD/89764/2012 to PO). AM acknowledges EMBO for a Short-Term Fellowship (EMBO ASTF 330-212). Transcript analysis was funded by NIH (grant P41GM103490) to KWM.
Abbreviations
- BabA
blood group antigen binding adhesin
- B3GNT5
β1,3-N-acetylglucosaminyltransferase 5
- B3GALT5
β1,3-galactosyltransferase 5
- FUT3
fucosyltransferase 3
- H. pylori
Helicobacter pylori
- LabA
LacdiNAc-specific adhesin
- Sialyl-Lea/x
sialyl-Lewis a/x
- SabA
sialic acid binding adhesin
- SNA
Sambucus nigra lectin
Footnotes
Contributors
AM and CAR conceived and designed research experiments and wrote the paper; RMP collected the gastric biopsies; experiments were performed by AM, AVN, MR, SN, DF, RMF, JG and MRS; LD, WX and FC performed histopathological and immunohistochemistry analysis; PO and CO performed the bioinformatics analysis; AM, AVN, LD, KWM and CAR analyzed the data; AVN, LD, CF, NTM, CO, MDR, FC and KWM contributed reagents, materials and revised the manuscript.
Transparency document
The Transparency document associated with this article can be found, in the online version.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbadis.2015.07.001.
References
- 1.Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007;133:659–672. doi: 10.1053/j.gastro.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012, Cancer Incidence and Mortality Worldwide, IARC CancerBase No 11. 1.0. International Agency for Researh on Cancer; Lyon, France: 2013. [Google Scholar]
- 3.Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hatakeyama M. Helicobacter pylori CagA and gastric Cancer: a paradigm for hit-and-run carcinogenesis. Cell Host & Microbe. 2014;15:306–316. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 6.Enroth H, Kraaz W, Engstrand L, Nyrén O, Rohan T. Helicobacter pylori strain types and risk of gastric cancer: a case–control study. Cancer Epidemiol Biomarkers Prev. 2000;9:981–985. [PubMed] [Google Scholar]
- 7.Magalhães A, Ismail MN, Reis CA. Sweet receptors mediate the adhesion of the gastric pathogen Helicobacter pylori: glycoproteomic strategies. Expert Rev Proteomics. 2010;7:307–310. doi: 10.1586/epr.10.18. [DOI] [PubMed] [Google Scholar]
- 8.Azevedo M, Eriksson S, Mendes N, Serpa J, Figueiredo C, Resende LP, Ruvoën-Clouet N, Haas R, Borén T, Pendu JL, David L. Infection by Helicobacter pylori expressing the BabA adhesin is influenced by the secretor phenotype. J Pathol. 2008;215:308–316. doi: 10.1002/path.2363. [DOI] [PubMed] [Google Scholar]
- 9.Ilver D, Arnqvist A, Ogren J, Frick I-M, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 10.Magalhaes A, Gomes J, Ismail MN, Haslam SM, Mendes N, Osorio H, David L, Le Pendu J, Haas R, Dell A, Boren T, Reis CA. Fut2-null mice display an altered glycosylation profile and impaired BabA-mediated Helicobacter pylori adhesion to gastric mucosa. Glycobiology. 2009;19:1525–1536. doi: 10.1093/glycob/cwp131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson K-A, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson K-E, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bäckström A, Lundberg C, Kersulyte D, Berg DE, Borén T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci U S A. 2004;101:16923–16928. doi: 10.1073/pnas.0404817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossez Y, Gosset P, Boneca IG, Magalhães A, Ecobichon C, Reis CA, Cieniewski-Bernard C, Joncquel Chevalier Curt M, Léonard R, Maes E, Sperandio B, Slomianny C, Sansonetti PJ, Michalski JC, Robbe-Masselot C. The LacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J Infect Dis. 2014;210:1286–1295. doi: 10.1093/infdis/jiu239. [DOI] [PubMed] [Google Scholar]
- 14.Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, Borén T, Haas R, Sasakawa C, Mimuro H. BabA-mediated adherence is a potentiator of the Helicobacter pylori Type IV secretion system activity. J Biol Chem. 2011;286:25256–25264. doi: 10.1074/jbc.M111.233601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandt S, Kwok T, Hartig R, König W, Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 17.Ota H, Nakayama J, Momose M, Hayama M, Akamatsu T, Katsuyama T, Graham DY, Genta RM. Helicobacter pylori infection produces reversible glycosylation changes to gastric mucins. Virchows Arch. 1998;433:419–426. doi: 10.1007/s004280050269. [DOI] [PubMed] [Google Scholar]
- 18.Pinho SS, Carvalho S, Marcos-Pinto R, Magalhães A, Oliveira C, Gu J, Dinis-Ribeiro M, Carneiro F, Seruca R, Reis CA. Gastric cancer: adding glycosylation to the equation. Trends Mol Med. 2013;19:664–676. doi: 10.1016/j.molmed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Colomb F, Krzewinski-Recchi MA, El Machhour F, Mensier E, Jaillard S, Steenackers A, Harduin-Lepers A, Lafitte JJ, Delannoy P, Groux-Degroote S. TNF regulates sialyl-Lewisx and 6-sulfo-sialyl-Lewisx expression in human lung through up-regulation of ST3GAL4 transcript isoform BX. Biochimie. 2012;94:2045–2053. doi: 10.1016/j.biochi.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 20.Colomb F, Vidal O, Bobowski M, Krzewinski-Recchi MA, Harduin-Lepers A, Mensier E, Jaillard S, Lafitte JJ, Delannoy P, Groux-Degroote S. TNF induces the expression of the sialyltransferase ST3Gal IV in human bronchial mucosa via MSK1/2 protein kinases and increases FliD/sialyl-Lewis(x)-mediated adhesion of Pseudomonas aeruginosa. Biochem J. 2014;457:79–87. doi: 10.1042/BJ20130989. [DOI] [PubMed] [Google Scholar]
- 21.Delmotte P, Degroote S, Lafitte J-J, Lamblin G, Perini J-M, Roussel P. Tumor necrosis factor α increases the expression of glycosyltransferases and sulfotransferases responsible for the biosynthesis of sialylated and/or sulfated Lewis x epitopes in the human bronchial mucosa. J Biol Chem. 2002;277:424–431. doi: 10.1074/jbc.M109958200. [DOI] [PubMed] [Google Scholar]
- 22.Marcos N, Magalhães A, Ferreira B, Oliveira M, Carvalho A, Mendes N, Gilmartin T, Head S, Figueiredo C, David L, Santos-Silva F, Reis C. Helicobacter pylori induces beta3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J Clin Invest. 2008;118:2325–2336. doi: 10.1172/JCI34324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcos-Pinto R, Carneiro F, Dinis-Ribeiro M, Wen X, Lopes C, Figueiredo C, Machado JC, Ferreira RM, Reis CA, Ferreira J, Pedroto I, Areias J. First-degree relatives of patients with early-onset gastric carcinoma show even at young ages a high prevalence of advanced OLGA/OLGIM stages and dysplasia. Aliment Pharmacol Ther. 2012;35:1451–1459. doi: 10.1111/j.1365-2036.2012.05111.x. [DOI] [PubMed] [Google Scholar]
- 24.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, Capelinha AF, Quint W, Caldas C, van Doorn L-J, Carneiro F, Sobrinho-Simões M. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 25.Lofling J, Holgersson J. Core saccharide dependence of sialyl Lewis X biosynthesis. Glycoconj J. 2009;26:33–40. doi: 10.1007/s10719-008-9159-z. [DOI] [PubMed] [Google Scholar]
- 26.Nairn AV, dela Rosa M, Moremen KW. Transcript analysis of stem cells. In: Minoru F, editor. Methods Enzymol. Vol. 479. Academic Press; 2010. pp. 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt S, Johnson N, Juettemann T, Kähäri AK, Keenan S, Kulesha E, Martin FJ, Maurel T, McLaren WM, Murphy DN, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ruffier M, Sheppard D, Taylor K, Thormann A, Trevanion SJ, Vullo A, Wilder SP, Wilson M, Zadissa A, Aken BL, Birney E, Cunningham F, Harrow J, Herrero J, Hubbard TJP, Kinsella R, Muffato M, Parker A, Spudich G, Yates A, Zerbino DR, Searle SMJ. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magalhães A, Marcos NT, Carvalho AS, David L, Figueiredo C, Bastos J, David G, Reis CA. Helicobacter pylori cag pathogenicity island-positive strains induce syndecan-4 expression in gastric epithelial cells. 2009 doi: 10.1111/j.1574-695X.2009.00569.x. [DOI] [PubMed] [Google Scholar]
- 30.Ferreira RM, Machado JC, Letley D, Atherton JC, Pardo ML, Gonzalez CA, Carneiro F, Figueiredo C. A novel method for genotyping the Helicobacter pylori vacA intermediate region directly in gastric biopsy specimens. J Clin Microbiol. 2012;50:3983–3989. doi: 10.1128/JCM.02087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Odenbreit S, Swoboda K, Barwig I, Ruhl S, Borén T, Koletzko S, Haas R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect Immun. 2009;77:3782–3790. doi: 10.1128/IAI.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes J, Magalhães A, Carvalho AS, Hernandez GE, Papp SL, Head SR, Michel V, David L, Gärtner F, Touati E, Reis CA. Glycophenotypic alterations induced by Pteridium aquilinum in mice gastric mucosa: synergistic effect with Helicobacter pylori infection. PLoS ONE. 2012;7:e38353. doi: 10.1371/journal.pone.0038353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindén S, Mahdavi J, Semino-Mora C, Olsen C, Carlstedt I, Borén T, Dubois A. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PloS Pathog. 2008;4:006–0013. doi: 10.1371/journal.ppat.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohno T, Vallström A, Rugge M, Ota H, Graham DY, Arnqvist A, Yamaoka Y. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J Infect Dis. 2011;203:726–735. doi: 10.1093/infdis/jiq090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gagneux P, Cheriyan M, Hurtado-Ziola N, van der Linden ECMB, Anderson D, McClure H, Varki A, Varki NM. Human-specific regulation of α2-6-linked sialic acids. J Biol Chem. 2003;278:48245–48250. doi: 10.1074/jbc.M309813200. [DOI] [PubMed] [Google Scholar]
- 37.Togayachi A, Akashima T, Ookubo R, Kudo T, Nishihara S, Iwasaki H, Natsume A, Mio H, Inokuchi J-i, Irimura T, Sasaki K, Narimatsu H. Molecular cloning and characterization of UDP-GlcNAc:lactosylceramide β1,3-N-acetylglucosaminyltransferase (β3Gn-T5), an essential enzyme for the expression of HNK-1 and Lewis X epitopes on glycolipids. J Biol Chem. 2001;276:22032–22040. doi: 10.1074/jbc.M011369200. [DOI] [PubMed] [Google Scholar]
- 38.Henion TR, Zhou D, Wolfer DP, Jungalwala FB, Hennet T. Cloning of a mouse β1,3 N-acetylglucosaminyltransferase GlcNAc(β1,3)Gal(β1,4)Glc-ceramide synthase gene encoding the key regulator of Lacto-series glycolipid biosynthesis. J Biol Chem. 2001;276:30261–30269. doi: 10.1074/jbc.M102979200. [DOI] [PubMed] [Google Scholar]
- 39.Togayachi A, Kozono Y, Ikehara Y, Ito H, Suzuki N, Tsunoda Y, Abe S, Sato T, Nakamura K, Suzuki M, Goda HM, Ito M, Kudo T, Takahashi S, Narimatsu H. Lack of lacto/neolacto-glycolipids enhances the formation of glycolipid-enriched micro-domains, facilitating B cell activation. Proc Natl Acad Sci U S A. 2010;107:11900–11905. doi: 10.1073/pnas.0914298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes EH, Ostrander GK, Clausen H, Graem N. Oncofetal expression of Lex carbohydrate antigens in human colonic adenocarcinomas. Regulation through type 2 core chain synthesis rather than fucosylation. J Biol Chem. 1987;262:11331–11338. [PubMed] [Google Scholar]
- 41.Holmes EH, Hakomori S, Ostrander GK. Synthesis of type 1 and 2 lacto series glycolipid antigens in human colonic adenocarcinoma and derived cell lines is due to activation of a normally unexpressed beta 1-3N-acetylglucosaminyltransferase. J Biol Chem. 1987;262:15649–15658. [PubMed] [Google Scholar]
- 42.Lin C-H, Fan Y-Y, Chen Y-Y, Wang S-H, Chen C-I, Yu L-C, Khoo K-H. Enhanced expression of β3-galactosyltransferase 5 activity is sufficient to induce in vivo synthesis of extended type 1 chains on lactosylceramides of selected human colonic carcinoma cell lines. Glycobiology. 2009;19:418–427. doi: 10.1093/glycob/cwn156. [DOI] [PubMed] [Google Scholar]
- 43.Salvini R, Bardoni A, Valli M, Trinchera M. β1,3-Galactosyltransferase β3Gal-T5 Acts on the GlcNAcβ1 → 3Galβ1 → 4GlcNAcβ1 → R sugar chains of carcinoembryonic antigen and other N-linked glycoproteins and is down-regulated in colon adenocar-cinomas. J Biol Chem. 2001;276:3564–3573. doi: 10.1074/jbc.M006662200. [DOI] [PubMed] [Google Scholar]
- 44.Mare L, Trinchera M. Suppression of β1,3galactosyltransferase β3Gal-T5 in cancer cells reduces sialyl-Lewis a and enhances poly N-acetyllactosamines and sialyl-Lewis x on O-glycans. Eur J Biochem. 2004;271:186–194. doi: 10.1046/j.1432-1033.2003.03919.x. [DOI] [PubMed] [Google Scholar]
- 45.Harduin-Lepers A, Krzewinski-Recchi MA, Colomb F, Foulquier F, Groux-Degroote S, Delannoy P. Sialyltransferases functions in cancers. Front Biosci (Elite Ed) 2012;4:499–515. doi: 10.2741/e396. [DOI] [PubMed] [Google Scholar]
- 46.Roche N, Ångström J, Hurtig M, Larsson T, Borén T, Teneberg S. Helicobacter pylori and complex gangliosides. Infect Immun. 2004;72:1519–1529. doi: 10.1128/IAI.72.3.1519-1529.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roche N, Larsson T, Ångström J, Teneberg S. Helicobacter pylori-binding ganglio-sides of human gastric adenocarcinoma. Glycobiology. 2001;11:935–944. doi: 10.1093/glycob/11.11.935. [DOI] [PubMed] [Google Scholar]
- 48.Wada A, Hasegawa M, Wong P-F, Shirai E, Shirai N, Tan L-J, Llanes R, Hojo H, Yamasaki E, Ichinose A, Ichinose Y, Senba M. Direct binding of gangliosides to Helicobacter pylori vacuolating cytotoxin (VacA) neutralizes its toxin activity. Glycobiology. 2010;20:668–678. doi: 10.1093/glycob/cwq014. [DOI] [PubMed] [Google Scholar]
- 49.Aspholm M, Olfat FO, Nordén J, Sondén B, Lundberg C, Sjostrom R, Altraja S, Odenbreit S, Haas R, Wadstrom T, Engstrand L, Semino-Mora C, Liu H, Dubois A, Teneberg S, Arnqvist A, Borén T. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2006;2:e110. doi: 10.1371/journal.ppat.0020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dohi T, Kawamura YI. Incomplete synthesis of the Sda/Cad blood group carbohydrate in gastrointestinal cancer. Biochim Biophys Acta. 2008;1780:467–471. doi: 10.1016/j.bbagen.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 51.Groux-Degroote S, Wavelet C, Krzewinski-Recchi MA, Portier L, Mortuaire M, Mihalache A, Trinchera M, Delannoy P, Malagolini N, Chiricolo M, Dall’Olio F, Harduin-Lepers A. B4GALNT2 gene expression controls the biosynthesis of Sda and sialyl Lewis X antigens in healthy and cancer human gastrointestinal tract. Int J Biochem Cell Biol. 2014;53:442–449. doi: 10.1016/j.biocel.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Kreisman LS, Cobb BA. Infection, inflammation and host carbohydrates: a Glyco-Evasion Hypothesis. Glycobiology. 2012;22:1019–1030. doi: 10.1093/glycob/cws070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basso D, Scrigner M, Toma A, Navaglia F, Di Mario F, Rugge M, Plebani M. Helicobacter pylori infection enhances mucosal interleukin-1 beta, interleukin-6, and the soluble receptor of interleukin-2. Int J Clin Lab Res. 1996;26:207–210. doi: 10.1007/BF02592984. [DOI] [PubMed] [Google Scholar]
- 55.Noach LA, Bosma NB, Jansen J, Hoek FJ, van Deventer SJ, Tytgat GN. Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand J Gastroenterol. 1994;29:425–429. doi: 10.3109/00365529409096833. [DOI] [PubMed] [Google Scholar]
- 56.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-[kappa]B signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 57.Lamb A, Chen L-F. The many roads traveled by Helicobacter pylori to NF-κB activation. Gut Microbes. 2010;1:109–113. doi: 10.4161/gmic.1.2.11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minami T, Miura M, Aird WC, Kodama T. Thrombin-induced autoinhibitory factor, down syndrome critical region-1, attenuates NFAT-dependent vascular cell adhesion molecule-1 expression and inflammation in the endothelium. J Biol Chem. 2006;281:20503–20520. doi: 10.1074/jbc.M513112200. [DOI] [PubMed] [Google Scholar]
- 59.Salama NR, Hartung ML, Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–399. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aberg A, Gideonsson P, Vallstrom A, Olofsson A, Ohman C, Rakhimova L, Boren T, Engstrand L, Brannstrom K, Arnqvist A. A repetitive DNA element regulates expression of the Helicobacter pylori sialic acid binding adhesin by a rheostat-like mechanism. PLoS Pathog. 2014;10:e1004234. doi: 10.1371/journal.ppat.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talarico S, Whitefield SE, Fero J, Haas R, Salama NR. Regulation of Helicobacter pylori adherence by gene conversion. Mol Microbiol. 2012;84:1050–1061. doi: 10.1111/j.1365-2958.2012.08073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodwin AC, Weinberger DM, Ford CB, Nelson JC, Snider JD, Hall JD, Paules CI, Peek RM, Forsyth MH. Expression of the Helicobacter pylori adhesin SabA is controlled via phase variation and the ArsRS signal transduction system. Microbiology. 2008;154:2231–2240. doi: 10.1099/mic.0.2007/016055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.