

Abstract
The blood-brain barrier (BBB) maintains brain homeostasis by limiting entry of substances to the central nervous system through interaction of transmembrane and intracellular proteins that make up endothelial cell tight junctions (TJs). Recently it was shown that the BBB can be modulated by disease pathologies including inflammatory pain. This study examined the effects of chronic inflammatory pain on the functional and molecular integrity of the BBB. Inflammatory pain was induced by injection of complete Freund’s adjuvant (CFA) into the right plantar hindpaw in female Sprague-Dawley rats under halothane anesthesia; control animals were injected with saline. Edema and hyperalgesia were assessed by plethysmography and infrared paw-withdrawal latency. At 72 h postinjection, significant edema formation and hyperalgesia were noted in the CFA-treated rats. Examination of permeability of the BBB by in situ perfusion of [14C]sucrose while rats were under pentobarbital anesthesia demonstrated that CFA treatment significantly increased brain sucrose uptake. Western blot analysis of BBB TJ proteins showed no change in expression of zonula occludens-1 (an accessory protein) or actin (a cytoskeletal protein) with CFA treatment. Expression of the transmembrane TJ proteins occludin and claudin-3 and -5 significantly changed with CFA treatment with a 60% decrease in occludin, a 450% increase in claudin-3, and a 615% increase in claudin-5 expression. This study demonstrates that during chronic inflammatory pain, alterations in BBB function are associated with changes in specific transmembrane TJ proteins.
Keywords: claudin, occludin, membrane permeability, diffusion, homeostasis, central nervous system, complete Freund’s adjuvant
The Blood-Brain Barrier (BBB) is critical for regulating and maintaining the brain microenvironment and thereby enabling proper neuronal function (8). It acts as both a physical and metabolic barrier, modulating the passage of materials from the peripheral circulation into the central nervous system (CNS). Without such tight regulation, the brain would be subject to fluctuating levels of circulating toxins, amino acids, and ions. Composing the anatomical basis of the BBB are the brain capillaries, which are characterized by a lack of fenestrations, the presence of tight junctional proteins that result in high transendothelial electrical resistances of 1,500–2,000 Ω/cm2, and multiple transport systems (2, 3, 6, 22). Each characteristic serves to limit the delivery of polar solutes and toxins as well as therapeutic compounds to the brain (30).
Tight junctions (TJs) span the apical region of the intercellular cleft of barrier tissues and allow for restriction of ion flux and paracellular diffusion. They are formed via a complex interaction of transmembrane, accessory, and cytoskeletal proteins. At one time thought to be static, TJs are actually dynamic structures regulated during both physiological and pathological states where protein expression changes can be associated with altered BBB paracellular permeability. The transmembrane proteins occludin and the claudins form the primary seal of the TJs. Several other proteins, including the zonula occludens (ZO) family and the actin cytoskeleton, are also involved in regulation of TJ integrity. These proteins have been demonstrated in numerous studies (1, 16) to be critical for maintenance of barrier integrity.
Such BBB integrity is compromised during many infections and disease states. Bacteria such as Vibrio cholera can cause alterations in TJ proteins that in turn cause opening of the BBB and subsequent CNS infection (18, 23, 24, 28, 31, 32). Ischemia leads to BBB disruptions and vasogenic edema via the opening of TJs, as do hypoxia and cerebral trauma (18, 23, 24, 28). In inflammatory CNS disorders such as multiple sclerosis, BBB alterations are often involved in disease development where increased permeability and lymphoid cell extravasation are early events (15, 19). Each of these diseases involves the disregulation of TJs that maintain the BBB.
Disruption of functional BBB permeability during inflammatory pain was previously demonstrated using inflammatory models where significant increases in [14C]sucrose (11, 13) and [3H]codeine (9) transport across the BBB were observed. Subsequent studies using direct intravenous injection of inflammatory agents [formalin, λ-carrageenan, complete Freund’s adjuvant (CFA)] showed no cytotoxic effects on microvascular endothelial cells or functional BBB permeability in vivo, which indicates that inflammation and/or pain can modulate both molecular and functional properties of the BBB (12, 13). Initial studies with CFA, a model of chronic inflammatory pain, demonstrated modulation of several TJ proteins including ZO-1, actin, and occludin. In this study, we extend those findings to investigate other TJ proteins (claudin-3 and -5) and protein localization. We also characterize permeability changes using the multiple time-uptake in situ perfusion technique. These data could have important implications for regulation of brain homeostasis during inflammation and pain.
MATERIALS AND METHODS
Radioisotopes, antibodies, and chemicals
The [14C]sucrose was purchased through ICN Pharmaceuticals (Irvine, CA) and had a specific activity of 492 mCi/mmol with >99.5% purity. Primary antibodies for ZO-1 (mouse), actin (mouse), occludin (rabbit), and claudin-1 (rabbit), -3 (rabbit), and -5 (mouse) were obtained from Zymed (San Francisco, CA). Conjugated anti-mouse IgG-horseradish peroxidase and anti-rabbit IgG-horseradish peroxidase were obtained through Amersham Life Science Products (Springfield, IL). CFA and all other chemicals, unless otherwise stated, were purchased from Sigma (St. Louis, MO).
Animals and treatments
All animal protocols were approved by the University of Arizona Institutional Animal Care and Use Committee and abide by the guidelines of the National Institutes of Health for the proper treatment of animals. Female Sprague-Dawley rats (Harlan; Indianapolis, IN) that weighed 250–300 g were housed under standard 12:12-h light-dark conditions and received food ad libitum. While rats were under halothane anesthesia, baseline paw-volume analysis was performed, and rats were injected subcutaneously with 100 μl of saline or CFA (at a 1:1 CFA-to-saline ratio for 50 μg) into the plantar surface of the right hindpaw. At 72 h postinjection, animals were assessed for edema and hyperalgesia and then anesthetized with pentobarbital sodium (64.7 mg/kg ip) for in situ brain perfusion or cerebral microvessel isolation.
Edema and behavioral tests
Edema was measured by the displacement of electrolyte solution in a plethysmometer (model 7141; Ugo Basile; Comerio VA, Italy) at preinjection and 24, 48, and 72 h postinjection. Hyperalgesia was measured by paw-removal latency upon exposure to infrared (IR) heat. The IR source heated linearly from 23.5°C (room temperature) at 0 s to 37°C at 15 s. The rats were habituated to individual boxes on an elevated glass table for 20 min before exposure of the right hindpaw plantar surface to a mobile IR source. Paw-withdrawal latencies were defined as the time(s) taken for the rat to remove its hindpaw from the heat source.
Mean blood pressure (MBP) was measured using a noninvasive blood pressure (BP) monitor (NIBP-8; Columbus Instruments; Columbus, OH). Briefly, animals were placed in a retaining cylinder with their tails under the warming unit (Columbus Instruments). They were then covered and allowed to habituate to their environment for 10 min. MBP was measured with the monitor a minimum of five times before data were recorded to allow the animal to become accustomed to a BP cuff. The animals were acclimated to the process for 4 days before hindpaw injection, and MBP was measured again 3 days postinjection.
In situ brain perfusion
Animals were anesthetized with pentobarbital sodium (64.7 mg/kg) and heparinized by injection (10,000 U/kg ip). At the neck, a ventral midline incision was made; the common carotid arteries were exposed and cannulated with silicone tubing while the jugular veins were cut to relieve pressure. The perfusion medium consisted of a Ringer solution (117 mM NaCl, 4.7 mM KCl, 0.8 mM MgSO4, 24.8 mM NaHCO3, 1.2 mM KH2PO4, 2.5 mM CaCl2, 10 mM D-glucose, 39 g/l dextran, 10 g/l BSA, pH 7.4) that contained Evans blue-labeled albumin. The solution was oxygenated with 95% O2-5% CO2 and passed via a peristaltic pump through a heating coil (37°C) and a bubble trap. Upon reaching 100 mmHg perfusion pressure and a flow rate of 3.1 ml/min, [14C]sucrose (10 mCi in 20 ml of Ringer solution) was infused (0.5 ml/min per hemisphere). The animals were perfused for 5, 10, or 20 min, after which they were decapitated, and their brains were harvested. The choroid plexus and meninges were removed, and the cerebral hemispheres were sectioned and homogenized. Samples of the radioactive perfusate from each carotid cannula were collected as a reference. Brain tissue and 100 μl of perfusate were prepared for liquid scintillation counting by incubation in 1 ml of TS-2 tissue solubilizer for 2 days, and radiation was counted after the addition of 100 μl of 30% acetic acid and 2.5 ml OptiPhase SuperMix liquid scintillation cocktail (PerkinElmer). Results are reported as the ratio of radioactivity in the brain to that in the perfusate (Rbr, measured in ml/g)
| (1) |
where Cbrain is the 14C measured in the brain (in dpm/g) and Cperfusate is the associated 14C measured in the perfusate (in dpm/ml). A least-squares regression of Rbr vs. perfusion time (t, measured in minutes) was performed for each experimental and control group
| (2) |
where VD is the initial volume of distribution of [14C]sucrose and Kin is the unidirectional transfer coefficient.
Microvessel isolation
While under halothane anesthesia, rats were decapitated and their brains were removed. Meninges and choroid plexuses were excised from the cerebral hemispheres, which were then homogenized in 6 ml of microvessel isolation buffer that contained (in mM) 103 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 15 HEPES, 2.5 NaHCO3, 10 D-glucose, and 1 sodium pyruvate with 10 g/l dextran (64,000 mol wt), pH 7.4, and a protease inhibitor cocktail that contained 0.2 mM phenylmethylsulfonyl fluoride, 1 mM benzamide, 1 mM NaVO4, 10 mM NaF, 10 mM sodium pyrophosphate, and 10 μg/ml each of aprotonin and leupeptin. We added 6 ml of 26% dextran, and the mixture was vortexed then centrifuged at 5,600 g for 10 min. The supernatant was aspirated and the pellets were resuspended in 10 ml of microisolation buffer before being passed through a 70-μm filter (Falcon; Becton-Dickinson; Franklin, NJ). The filtered homogenates were centrifuged at 3,000 g for 10 min at 4°C. Protein was extracted from the pellets with 6 M urea lysis buffer (6 M urea, 0.1% Triton X-100, 10 mM Tris·HCl, pH 8.0, 1 mM dithiothreitol, 5 mM MgCl2, 5 mM EGTA, and 150 mM NaCl) with the protease inhibitor cocktail. Protein concentrations were determined by bicinchoninic acid protein assay (Pierce; Rockford, IL).
TJ protein analysis
Protein isolated from microvessels was analyzed for expression of the TJ proteins ZO-1, actin, occludin, and claudin-1, -3, and -5. Microvessel protein samples (40 μg) were resolved on a 4–12% Tris-glycine gel (Novex; San Diego, CA) for 1.5 h at 125 V and transferred to a nitrocellulose membrane for 45 min at 240 mA. GelCode Blue stain reagent (Pierce) was used to ensure proper loading of protein. The nitrocellulose membranes were incubated with blocking buffer (20 mM Tris base, 137 mM NaCl, 2M HCl, 0.1% Tween 20; pH 7.6) with 5% nonfat milk overnight at 4°C. Blots were incubated at room temperature for 1.5 h with primary antibody (1:1,000 dilution), washed in blocking buffer with 5% nonfat milk at room temperature for 1 h, and incubated with secondary antibody (1:1,500 dilution) for 45 min at room temperature. Blots were developed using enhanced chemiluminescence (ECL+; Amersham Life Science Products) and analyzed using Scion Image software (Scion; Frederick, MD).
Immunofluorescence microscopy
Microvessels isolated as described above were smeared onto microscope slides and heat-fixed at 95°C for 10 min. After fixation (with 3.7% formaldehyde in PBS for 10 min) and permeabilization (with 0.1% Triton X-100 in PBS for 5 min), the vessels were blocked for 30 min in PBS with 1% BSA. Slides were incubated with 1:100 dilutions of primary antibodies (ZO-1, occludin, and claudin-1, -3, and -5) in PBS with 1% BSA for 30 min and were rinsed and reblocked for 30 min with 1% BSA in PBS. Slides were then incubated with Alexa Fluor 488-conjugated anti-rabbit or anti-mouse IgG (R&D Systems; Minneapolis, MN) diluted in 1% BSA in PBS for 30 min (at 1:500 dilutions except for claudin-3, which was a 1:1,000 dilution). Vessels from saline- and CFA-treated animals were stained simultaneously for each protein. After placement and sealing of coverslips with Vectashield, photographs were taken with ×100 oil-immersion objectives on a Nikon TE-300 fluorescence microscope with a fluorescein filter.
Statistical analysis
For the in situ brain perfusion experiments, statistical comparisons of the regression coefficients Kin and VD were performed as previously described (11) in accordance with the methods of Glantz (8). Analysis of all other data was performed using Student’s t-test.
RESULTS
Edema, hyperalgesia, and BP
To confirm proper injection of CFA, paw volume was measured pre- and postinjection in both the injected paw and the paw contralateral to the injection. No increases in paw volume were noted with saline injection in either paw or in the contralateral paws of CFA-treated rats. Significant (P < 0.001) edema was noted in the CFA-injected hindpaw over time. The injected hindpaw increased in volume by 84.5 ± 5.8, 76.8 ± 7.3, and 60.8 ± 4.9% at 24, 48, and 72 h, respectively (Fig. 1).
Fig. 1.
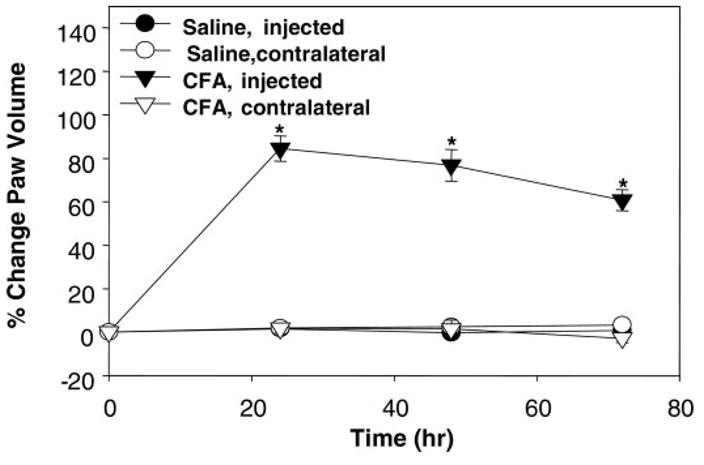
Edema established with injection of complete Freund’s adjuvant (CFA). Percent change in paw volume at 24, 48, and 72 h postinjection of saline or CFA. CFA caused significant edema at each time measured in only the ipsilateral paw compared with saline; *P < 0.001. For comparison purposes, the contralateral paw of the CFA-injected animal as well as both paws of the saline-injected animal were also measured and demonstrated no volume change; n ≥ 20 rats/treatment.
Hyperalgesia was measured as a decrease in the time (in seconds) taken to remove a paw from an IR heat source. In the saline-injected right hindpaw, the paw removal latency times were 9.8 ± 0.6, 9.6 ± 0.4, and 10.3 ± 0.6 s at 24, 48, and 72 h, respectively. Upon CFA injection, a significant decrease (P < 0.001) in the removal latency was observed at all time points measured. The paw was removed from the IR heat source by 3.1 ± 0.5, 4.6 ± 1.3, and 5.1 ± 0.3 s at 24, 48, and 72 h, respectively (Fig. 2). Exclusion criteria were set on paw-removal latency (in seconds) as follows: an animal within a treatment group whose removal latency was outside of the parameters average (in seconds) ±2 standard deviations and any CFA-treated animal whose paw removal latency was greater than the average of saline-treated animals minus 2 standard deviations. Two CFA-treated animals were removed from the in situ brain perfusion study, and three CFA-treated animals were removed from the TJ protein analysis by these criteria.
Fig. 2.
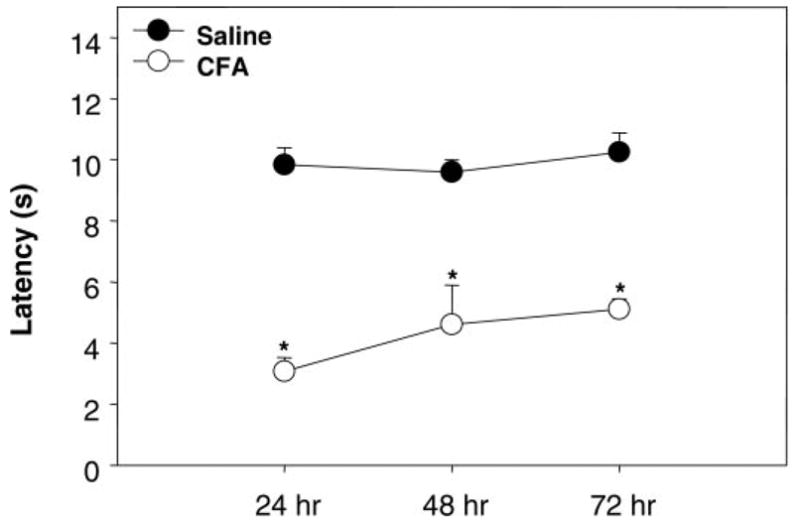
Hyperalgesia established with CFA. Hyperalgesia was measured as paw-removal latency (in seconds) from infrared heat source. At 24, 48, and 72 h postinjection, CFA caused a significant decrease in removal latency compared with saline; *P < 0.001; n ≥ 20 rats/treatment.
To ensure that any potential changes in either TJ expression or permeability were not due to changes in BP, we investigated the effects of saline and CFA injections on awake-state MBP. There were no significant differences between pre- and postinjection MBP values for saline (114.1 ± 3 vs. 126.2 ± 6.4 mmHg) or for CFA (118.5 ± 4.1 vs. 116.2 ± 2.3 mmHg).
In situ brain perfusion
We examined the functional integrity of the BBB by perfusing the brain with [14C]sucrose for 5, 10, or 20 min. Visualization of the whole brain immediately after perfusion showed no noticeable parenchymal leakage of the Evans blue albumin. CFA treatment for 72 h led to a significant (P < 0.05) increase in the Kin coefficient compared with saline treatment. Kin was increased from 0.0 ± 0.3 to 0.6 ± 0.1 μl·g−1·min−1 (Fig. 3 and Table 1). Estimated initial vascular VD values were 8.2 ± 1.2 and 14.8 ± 3.7 μl/g with no significant difference between treatment groups (Table 1).
Fig. 3.
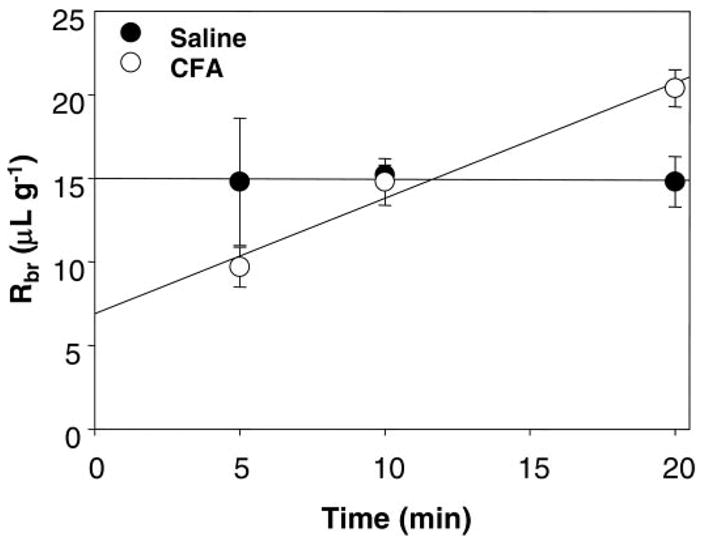
Multiple time [14C]sucrose uptake. In this study, [14C]sucrose was used as a marker of blood-brain barrier paracellular permeability. Values are mean brain radioactivity (Rbr) ± SE; n = 4–8 rats/time point. CFA significantly increased the rate of [14C]sucrose uptake into the brain (Kin) without significantly altering its initial vascular distribution (VD).
Table 1.
Analyses from multitime-uptake parameters
Values are extrapolated regression coefficients ± SE;
n = 14 or
n = 18 rats. Kin, rate of [14C]sucrose uptake; VD, initial volume of distribution. Kin and VD are regression coefficients calculated from the plot in Fig. 3. Complete Freund’s adjuvant (CFA) treatment significantly increased the rate of brain [14C]sucrose uptake compared with saline.
P < 0.05. There was no change in initial VD with CFA treatment.
Expression of TJ proteins
We measured the expression of the TJ proteins ZO-1, occludin, actin, and claudin-1, -3, and -5 from isolated brain microvessels. We did not detect any claudin-1 in microvessel preparations (data not shown). After 72 h, CFA did not induce any changes in ZO-1 (100 ± 11 vs. 83 ± 12%) or actin (100 ± 17 vs. 117 ± 19%) expression. There were alterations in each of the transmembrane TJ proteins examined, which included occludin and claudin-3 and -5. Occludin was significantly (P = 0.001) decreased from 100 ± 14 to 40 ± 9% in the CFA-injected rats by 72 h. Also, at 72 h post-CFA injection, expression of both claudin-3 and -5 was significantly (P < 0.001) increased greater than five- and seven-fold, respectively (Fig. 4), from 100 ± 9 to 550 ± 71% for claudin-3 and from 100 ± 14 to 715 ± 138% for claudin-5.
Fig. 4.
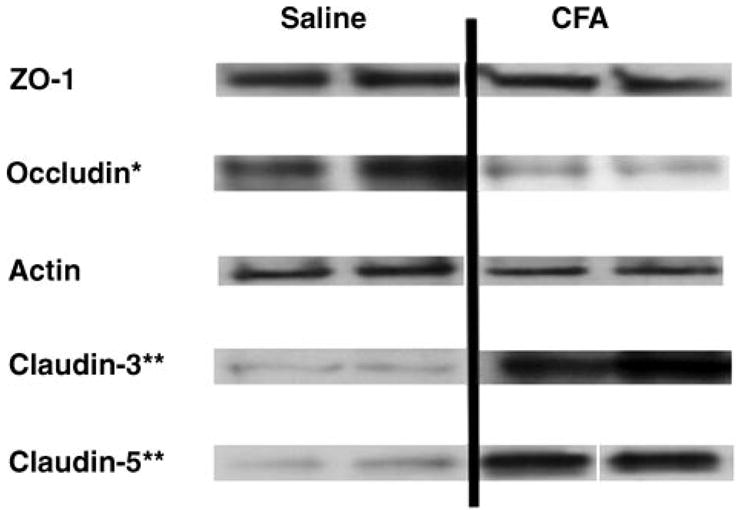
Tight junction (TJ) protein expression. Expression of various TJ proteins was measured 72 h post-CFA injection (n ≥ 6 rats). CFA was not found to alter the expression of the accessory TJ protein zonula occludens (ZO)-1 or the cytoskeletal support protein actin. Western blots showing protein expression demonstrate representative results from at least triplicate independent experiments. Optical density of each protein band was measured and normalized to saline control expression on each membrane. Expression of the transmembrane protein occludin was significantly decreased (*P = 0.001). Expression of both claudin-3 and -5 was significantly increased (**P < 0.001). Gels were stained with Coomassie blue as a loading control.
Localization of transmembrane TJ proteins
We also examined the localization of the TJ proteins occludin and claudin-3 and -5. With the use of fluorescence microscopy, localization was qualitatively assessed, and no alteration in the pattern of localization of either claudin-3 or -5 (Fig. 5) was noted. There was, however, a change noted in the localization of occludin from a continuous, marginal distribution to a more punctate pattern of immunoreactivity (Fig. 5, white arrows).
Fig. 5.
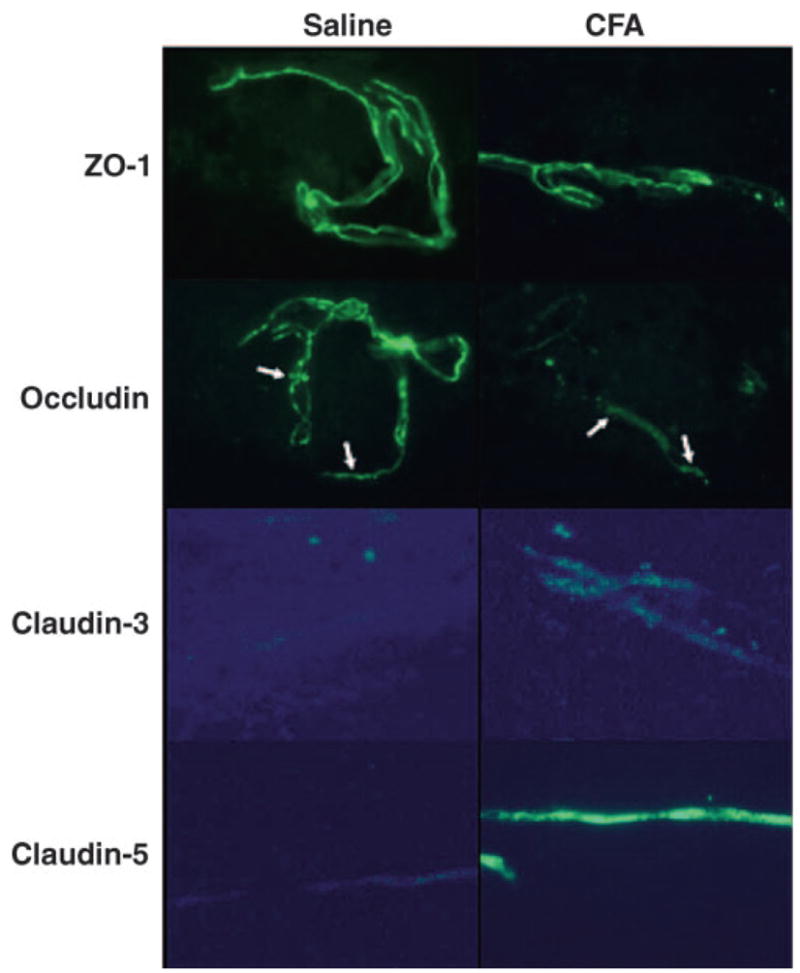
Localization of TJ proteins. Representative micrographs of changing blood-brain barrier TJ proteins occludin and claudin-3 and -5; n ≥ 6 rats. Data represent minimally triplicate independent and separate experiments. No changes in localization were noted with claudin-3 or -5. Occludin, however, was noted to change from a continuous localization to a more punctate distribution (white arrows).
DISCUSSION
The integrity of the BBB is critical for maintaining the homeostatic environment of the brain. In this study, we have examined the functional and molecular effects of CFA-induced chronic inflammatory pain on the BBB TJs and have demonstrated a change in paracellular permeability correlating with significant alterations in key transmembrane proteins. This study confirms previous evidence that CFA has a significant effect on BBB integrity (14, 20) and extends our understanding of the molecular mechanisms associated with changes in permeability.
We demonstrated significant changes in both paw edema formation and hyperalgesia caused by CFA and examined paracellular permeability under conditions of CFA-induced chronic inflammatory pain. Changes in cerebral hemodynamics and BP have previously been shown to modulate the BBB (17, 23). To ensure that these factors were not involved in our model, we investigated MBP in saline- and CFA-treated animals and found no significant change. This agrees with our previous data, which showed no significant effect on cerebral blood flow of CFA-treated animals (14). These studies indicate that changes in cerebral hemodynamics do not play a significant role in the observed changes in TJs.
The multiple time-uptake studies of [14C]sucrose show a significant increase in Kin (i.e., rate of sucrose uptake) with no significant increase in initial VD. This supports the data (see RESULTS), which show that the increased sucrose content of the brain is due to an increase in transport rather than a change in vascular volume. Previously, we saw no change in sucrose trapping in endothelial cells (14), which indicated that the increased sucrose transport is indeed via a paracellular route.
Because TJs are key to BBB paracellular integrity, we followed this observation with the examination of specific TJ proteins with isolated microvessels from saline- and CFA-treated animals. No changes were noted in the accessory and cytoskeletal proteins ZO-1 and actin. Significant alterations, however, were noted in each of the transmembrane proteins of the BBB including occludin and claudin-3 and -5. Occludin decreased by 60% with CFA treatment, whereas claudin-3 and -5 increased more than 450 and 600%, respectively. We further demonstrated that occludin localization is changed from continuous to punctate. Our data are the first to correlate a change in BBB paracellular permeability with alterations in TJ transmembrane proteins after CFA-induced chronic inflammatory pain insult.
Previously, CFA treatment was shown to reduce occludin content significantly (14). In this study, we have shown the same response using both Western blot analysis and fluorescence microscopy. The occludin localization changed from a marginal distribution to a more punctuate staining pattern in CFA-treated animals. In contrast with the previous studies (14), we saw no significant increases in ZO-1 or actin protein expression. The previous CFA studies used Triton X-100 to isolate protein. Studies indicate that TJs are micromembrane domains (19) that have both Triton X-soluble and -insoluble components. The urea buffer used in our study will isolate protein from both of these fractions. Therefore, the differences between ZO-1 and actin seen in the earlier study (14) and this investigation are due to movement from Triton X-insoluble to Triton X-soluble (i.e., membrane-associated to cytoplasmic) fractions. The fluorescence microscopy studies indicate that these fractions are still closely associated with the TJ area, as no change in gross localization was observed.
The claudin family contains over 20 members that are characterized by four membrane-spanning domains, two extra-cellular loops, and two cytoplasmic termini. At the BBB, claudin-1, -3, and -5 have been reported (1, 22). In our experiments, by both Western blot analysis and fluorescence microscopy, we were unable to visualize claudin-1 expression. This has been noted by other groups examining microvessels isolated from rat brains (11). The majority of the literature indicates a correlation between reductions in claudin-3 and -5 and decreased BBB permeability (15, 16, 25, 26). Our data, conversely, demonstrate increases in claudin-3 and -5 expression in an in vivo model of inflammatory pain. In vitro work has been done examining the effect of claudin overexpression in lung epithelium (5). Transfection of NIH/3T3 cells with claudin-3 resulted in increased transendothelial electrical resistance and decreased permeability. This was completely reversed when cotransfection of claudin-5 was performed, which indicates that claudin-5 overexpression reverses permeability changes demonstrated by claudin-3 (5). Other studies (4) have shown that the levels of claudin-3 and -5 are vital for regulating TJ permeability. Overall, this demonstrates the complexity of the TJ and the intricate balance of the protein levels and interactions between TJ proteins that is critical for maintaining BBB integrity.
We have shown that chronic inflammatory pain affects both the functional and molecular properties of the BBB. Moreover, we demonstrate a correlation between increased BBB permeability and altered expression of key transmembrane TJ proteins responsible in maintaining BBB integrity. Our results suggest that peripheral chronic inflammatory pain leads to a reorganization of the TJ proteins and thus to increased paracellular diffusion. These changes may have a significant effect on the delivery of therapeutic compounds to the brain under pathological conditions. Clinical dosing regimens may need reevaluation in light of these findings of increased paracellular permeability under chronic inflammatory pain where dose escalation is considered during pathological states and CNS toxicity is a concern.
Acknowledgments
GRANTS
This work was supported by National Institutes of Health Grants R01 NS-039592, NS-042652, DA-011271, and HL-07249.
References
- 1.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 2.Brightman MW, Kadota Y. Nonpermeable and permeable vessels of the brain. NIDA Res Monogr. 1992;120:87–107. [PubMed] [Google Scholar]
- 3.Butt AM. Effect of inflammatory agents on electrical resistance across the blood-brain barrier in pial microvessels of anaesthetized rats. Brain Res. 1995;696:145–150. doi: 10.1016/0006-8993(95)00811-4. [DOI] [PubMed] [Google Scholar]
- 4.Chen SP, Zhou B, Willis BC, Sandoval AJ, Liebler JM, Kim KJ, Ann DK, Crandall ED, Borok Z. Effects of transdifferentiation and EGF on claudin isoform expression in alveolar epithelial cells. J Appl Physiol. 2005;98:322–328. doi: 10.1152/japplphysiol.00681.2004. [DOI] [PubMed] [Google Scholar]
- 5.Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166–L1178. doi: 10.1152/ajplung.00182.2003. [DOI] [PubMed] [Google Scholar]
- 6.Crone C, Christensen O. Electrical resistance of a capillary endothelium. J Gen Physiol. 1981;77:349–371. doi: 10.1085/jgp.77.4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glantz S. Primer of Biostatistics. New York: McGraw Hill; 2002. [Google Scholar]
- 8.Goldstein GW, Betz AL. The blood-brain barrier. Sci Am. 1986;255:74–83. doi: 10.1038/scientificamerican0986-74. [DOI] [PubMed] [Google Scholar]
- 9.Hau VS, Huber JD, Campos CR, Davis RT, Davis TP. Effect of lambda-carrageenan-induced inflammatory pain on brain uptake of codeine and antinociception. Brain Res. 2004;1018:257–264. doi: 10.1016/j.brainres.2004.05.081. [DOI] [PubMed] [Google Scholar]
- 10.Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, Davis TP. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004;1027:48–58. doi: 10.1016/j.brainres.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 11.Huber JD, Hau VS, Borg L, Campos CR, Egleton RD, Davis TP. Blood-brain barrier tight junctions are altered during a 72-h exposure to lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol. 2002;283:H1531–H1537. doi: 10.1152/ajpheart.00027.2002. [DOI] [PubMed] [Google Scholar]
- 12.Huber JD, Hau VS, Mark KS, Brown RC, Campos CR, Davis TP. Viability of microvascular endothelial cells to direct exposure of formalin, lambda-carrageenan, and complete Freund’s adjuvant. Eur J Pharmacol. 2002;450:297–304. doi: 10.1016/s0014-2999(02)02150-7. [DOI] [PubMed] [Google Scholar]
- 13.Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2001;280:H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- 14.Kanda T, Numata Y, Mizusawa H. Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. J Neurol Neurosurg Psychiatry. 2004;75:765–769. doi: 10.1136/jnnp.2003.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201:319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- 16.Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EJ, Hung YC, Lee MY. Early alterations in cerebral hemodynamics, brain metabolism, and blood-brain barrier permeability in experimental intracerebral hemorrhage. J Neurosurg. 1999;91:1013–1019. doi: 10.3171/jns.1999.91.6.1013. [DOI] [PubMed] [Google Scholar]
- 18.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H1485–H1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131–141. doi: 10.1227/01.neu.0000097715.11966.8e. [DOI] [PubMed] [Google Scholar]
- 20.Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- 21.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund’s adjuvant in mice provoke leakage of serum proteins through the blood-brain barrier without inducing reactive gliosis. Brain Res. 1999;832:84–96. doi: 10.1016/s0006-8993(99)01479-1. [DOI] [PubMed] [Google Scholar]
- 22.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999;42:209–216. doi: 10.1016/s0033-0620(99)70003-4. [DOI] [PubMed] [Google Scholar]
- 24.Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2000;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells. 1998;3:569–573. doi: 10.1046/j.1365-2443.1998.00212.x. [DOI] [PubMed] [Google Scholar]
- 26.Ueno M, Sakamoto H, Liao YJ, Onodera M, Huang CL, Miyanaka H, Nakagawa T. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem Cell Biol. 2004;122:131–137. doi: 10.1007/s00418-004-0684-y. [DOI] [PubMed] [Google Scholar]
- 27.Willis CL, Leach L, Clarke GJ, Nolan CC, Ray DE. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia. 2004;48:1–13. doi: 10.1002/glia.20049. [DOI] [PubMed] [Google Scholar]
- 28.Witt KA, Mark KS, Hom S, Davis TP. Effect of hypoxia/reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285:H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 29.Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol (Berl) 2003;105:586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 30.Wolka AM, Huber JD, Davis TP. Pain and the blood-brain barrier: obstacles to drug delivery. Adv Drug Deliv Rev. 2003;55:987–1006. doi: 10.1016/s0169-409x(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 31.Wu Z, Milton D, Nybom P, Sjo A, Magnusson KE. Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb Pathog. 1996;21:111–123. doi: 10.1006/mpat.1996.0047. [DOI] [PubMed] [Google Scholar]
- 32.Wu Z, Nybom P, Magnusson KE. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol. 2000;2:11–17. doi: 10.1046/j.1462-5822.2000.00025.x. [DOI] [PubMed] [Google Scholar]