

Abstract
The fusion of echinoderm microtubule-associated protein-like 4 with the anaplastic lymphoma kinase (EML4-ALK) is found in 3%–7% of non-small-cell lung cancer (NSCLC) cases and confers sensitivity to crizotinib, the first United States Food and Drug Administration (FDA)-approved ALK inhibitor drug. Although crizotinib has an excellent initial therapeutic effect, acquired resistance to this drug invariably develops within the first year of treatment. Resistance may involve secondary gatekeeper mutations within the ALK gene interfering with crizotinib–ALK interactions, or compensatory activation of aberrant bypass signaling pathways. New therapeutic strategies to overcome crizotinib resistance are needed. Ceritinib, a second-generation ALK inhibitor, overcomes several crizotinib-resistant ALK mutations and has demonstrated efficacy against tumor growth in several in vitro and in vivo preclinical models of crizotinib resistance. Notably, the dose-escalation Phase I ASCEND-1 trial has shown a marked activity of ceritinib in both crizotinib-naïve and crizotinib-resistant ALK-rearranged lung cancer. The overall response rate was 58% in a subgroup of patients with ALK-rearranged late-stage NSCLC. Drug discontinuation rate due to toxicity was 10%. The standard dose was established at 750 mg daily. This paper outlines the pathogenesis and treatment of ALK-positive lung cancer, focuses on the preclinical and clinical results surrounding the accelerated FDA approval of ceritinib for the treatment of ALK-positive metastatic NSCLC patients who have progressed on/or are crizotinib intolerant, and discusses the potential efforts seeking to maximize ceritinib efficacy and expand its usage to other indications in cancer therapy.
Keywords: crizotinib, EML4-ALK, ceritinib, lung cancer, targeted therapy, personalized medicine
Introduction
Non-small-cell lung cancer (NSCLC) remains the leading cause of cancer deaths in the USA and claims more lives each year than all other major cancers combined.1 NSCLC is generally diagnosed at an advanced and incurable stage, at which time the median overall survival is measured in months. While 24%–30% of patients with advanced NSCLC respond initially to chemotherapy, most responders relapse within 6 months.2–4 With the dawn of personalized medicine, the discovery of various genetic alterations that promote cancer growth and survival, such as epidermal growth factor receptor (EGFR) mutations and echinoderm microtubule-associated protein like 4-anaplastic lymphoma kinase (EML4-ALK) rearrangements, have revolutionized treatment paradigms for patients with lung cancer.5 In fact, the past few years have witnessed the emergence of multiple targeted therapies directed against EGFR mutation, including gefitinib, erlotinib, and afatinib, or against ALK rearrangement such as crizotinib and more recently ceritinib.
About 3%–7% of metastatic NSCLCs (mNSCLC) carry the EML4-ALK oncogene fusion rearrangement. In this subset population, the malignancy is highly sensitive to therapy with ALK-targeted inhibitors.6,7 Indeed, crizotinib improves progression-free survival (PFS) by 4.7 months when compared with standard cytotoxic chemotherapy in previously treated patients with ALK-positive mNSCLC.8 This finding represents a major breakthrough in NSCLC precision medicine. Unfortunately, despite initial responses, almost all ALK-rearranged lung cancer patients relapse within 12 months of treatment initiation, secondary to acquired drug resistance. While the treatment options after failure to crizotinib are limited, the advent of new generation ALK inhibitors, notably ceritinib, has broadened the therapeutic arsenal for ALK-rearranged lung cancer. Ceritinib is a second-generation ALK inhibitor, which is 20 times more potent than crizotinib and has demonstrated clinical efficacy in patients with ALK-positive mNSCLC who have failed therapy with crizotinib.9
In this review, we briefly discuss the biology and treatment of ALK-positive mNSCLC. We summarize the molecular basis and the current strategies to overcome crizotinib resistance. In particular, we focus on the preclinical experimental studies, the pharmacology and clinical trials that have surrounded the accelerated United States Food and Drug Administration (FDA) approval of ceritinib. Finally, we touch upon the future potential use of ceritinib in other cancer indications and the unmet needs in the treatment of ALK-rearranged lung cancer.
Molecular basis and pathogenesis of ALK-positive lung cancer
EML4-ALK was identified as a transforming lung cancer fusion gene in 2007.7 This genetic rearrangement accounts for 3%–7% of NSCLC cases, with a higher prevalence among younger individuals, patients with adenocarcinoma, and nonsmokers.10 The ALK is a transmembrane tyrosine kinase receptor whose expression is restricted to certain tissues, such as neural tissue, the small intestine, and testes, and is thought to function in central nervous system (CNS) development.11 ALK is normally activated by ligand binding to the extracellular receptor domain, leading to dimerization and receptor activation.7 The EML4-ALK fusion oncogene arises from an inversion on the short arm of chromosome 2 (Inv(2)(p21p23)) that juxtaposes exons 1–13 of EML4 to exons 20–29 of ALK.7 Multiple variants of EML4-ALK have been described that encode the same cytoplasmic tyrosine kinase domain of ALK with different truncations of EML4.12,13
Fusion of the ALK and EML4 genes results in the replacement of the extracellular and transmembrane portions of the ALK protein with portions of the EML4 protein.12,13 The resulting chimeric EML4-ALK protein is an EML4 N-terminus and ALK C-terminus domain-derived fusion protein that functions as an intracellular tyrosine kinase. Unlike the normal ALK protein located on the cell surface, the abnormal EML-ALK fusion protein locates to the cytoplasm. The amino-coiled coil portion of EML4 causes the fusion protein to be constitutively activated without the need of ligand binding. Consequently, the activation of multiple downstream signaling pathways (primarily the PI3K/mTOR and RAS/RAF/MAPK) by the aberrant ALK protein leads to cell transformation and uncontrolled cancer cell proliferation.14–16 Tyrosine kinase inhibitors that target the ALK kinase activity have powerful antiproliferative effects when administered to EML4-ALK-positive lung cancer patients.8,9
Crizotinib: first FDA-approved ALK inhibitor drug
Crizotinib antagonizes the tyrosine kinase activity of ALK by competing with adenosine triphosphate (ATP) for binding to the ALK ATP pocket. It was the first ALK inhibitor to be approved by the FDA for the treatment of patients with ALK-positive advanced NSCLC. This molecule is associated with effective antitumor activity in both previously chemotherapy-treated and chemotherapy-naïve patients. In a randomized, open-label Phase III clinical trial (PROFILE 1007),8 347 patients with ALK-positive NSCLC previously treated with chemotherapy were randomized to crizotinib or to chemotherapy with either docetaxel or pemetrexed. Crizotinib has achieved a significantly higher response rate and longer PFS compared with chemotherapy. The overall response rate was 65% in the crizotinib group and 20% in the chemotherapy group (P<0.001). The median PFS was 7.7 months for patients treated with Crizotinib versus 3 months in the chemotherapy arm (HR 0.49, 95% CI: 0.37–0.64). However, an interim overall survival analysis showed no significant improvement with crizotinib as compared with the chemotherapy arm.
In another Phase III randomized clinical trial (PROFILE 1014),17 343 chemotherapy naïve patients with ALK-positive NSCLC were randomly assigned to crizotinib or chemotherapy (pemetrexed combined with either carboplatin or cisplatin). PFS was significantly prolonged with crizotinib in comparison with chemotherapy (10.9 months versus 7.0 months, HR 0.45, 95% CI: 0.35–0.60). Objective response rates were 74% and 45%, respectively (P<0.001). No overall survival difference was noticed. The absence of overall survival benefit in both trials may be due to the fact that crossover to crizotinib was permitted upon progression to chemotherapy in both studies. The National Comprehensive Cancer Network recommends that patients with advanced ALK-positive NSCLC receive crizotinib as a first-line treatment. For patients who develop crizotinib resistance or who are unable to tolerate crizotinib, treatment with ceritinib is recommended.18
Pathophysiology and therapeutic strategies to overcome crizotinib resistance
While crizotinib is highly effective in patients with ALK-positive NSCLC, ultimately all the patients develop drug resistance, typically after an average of 12 months following treatment initiation.19 Acquired crizotinib resistance may be the result of either pharmacological or biological mechanisms. Pharmacological resistance is secondary to inadequate drug bioavailability. Development of brain metastasis while on crizotinib is a typical example of this kind of resistance, since crizotinib has very low CNS penetration. The biological mechanisms of resistance can be broadly categorized into two classes: ALK dominant and ALK nondominant.19–21 ALK-dominant resistance mechanisms accounts for approximately one-third of the cases. It involves either an ALK fusion gene amplification or an acquired secondary mutation located within the ALK tyrosine kinase domain, sterically interfering with the ability of the drug to bind and block the tyrosine kinase domain. The most frequently identified mutations are the L1196M mutation, followed by the G1269A mutation.22,23 Among the other known ALK secondary mutations detected in crizotinib-resistant patients are 1151T-ins, L1152R, C1156Y, G1202R, and S1206Y.24–26 While second-generation ALK inhibitors with improved potency and selectivity are effective against most of the secondary mutation of ALK protein, the G1202R mutation confers resistance to these new drugs, including ceritinib.25
ALK-nondominant mechanisms of resistance are secondary to compensatory changes within the targeted pathway that bypass crizotinib-mediated inhibition. It accounts for approximately one-third of crizotinib-resistant cases. These driver pathways include insulin-like growth factor-1 receptor, EGFR, KIT, c-MET, KRAS, and the mTOR pathways.22–25 Combination therapies employing inhibitors of these bypass-signaling pathways may overcome crizotinib resistance.26 Importantly, strong preclinical data showed that hsp90 inhibitors are able to overcome resistance to crizotinib.27 This has provided a rationale to test their combinatorial activity in clinic. In the remaining one-third of cases, the mechanisms of crizotinib resistance are presently unknown22,26 (Table 1). From a practical standpoint, a rebiopsy at the time of progression may be critical to delineate the underlying mechanisms of crizotinib failure and subsequently guide the clinician in choosing the appropriate therapy. When resistance is driven by an ALK-dominant mechanism, a second-generation ALK inhibitor may potentially overcome refractoriness to crizotinib. In ALK-nondominant refractory tumors, approaches combining crizotinib with targeted agents directed against the activated escape prosurvival pathway may be appropriate in restoring sensitivity to crizotinib. Interestingly, clinical benefits were observed in patients with acquired resistance to crizotinib when treated with ceritinib, regardless of whether a resistance ALK mutation was detected. This suggests that ceritinib may be targeting these aberrant escape pathways as well. Nevertheless, an approach based on rebiopsy at time of progression needs to be validated by clinical trials.20
Table 1.
Mechanisms of crizotinib resistance
| Mechanism | Example | Comments |
|---|---|---|
| Secondary mutation in the ALK gene | L1196M, G1202R, G1269A, L1152R, C1156Y, F1174L, G1206Y, 1151Tins | L1196M and G1269A are the two most common mutations and confer a sensitivity to ceritinib, whereas G1202R is refractory to ceritinib. |
| ALK gene amplification | ||
| Activation of an alternative escape pathway | EGFR mutation or amplification(br)c-KIT amplification(br)KRAS mutation(br)MET amplification | Studies evaluating ALK inhibitor in combination with agents that suppress these bypass signaling pathways are needed. |
Review of pharmacology, mode of action, efficacy, and tolerability of ceritinib
Chemistry of ceritinib
Prior to the discovery of ceritinib, researchers designed numerous ALK inhibitors, among which TAE684 was found to have a potent ALK kinase inhibitory property (Figure 1A). However, this molecule was found to form various reactive adducts that may play a role in serious adverse events, making this drug a nonviable clinic option.28 Initial work focusing on improving the kinase selectivity of TAE684 led to synthesis of compound 7, which was constructed by replacing the methoxy moiety with the isopropoxy moiety at the aniline ring. This modification confers a higher affinity for the second-site-mutated ALK. Similar to compound TAE684, reactive metabolite formation was a problem in advancing this drug into the clinical arena. Further structural modifications of compound 7 have ultimately led to the discovery of LDK378 (ceritinib, Figure 1B). The rationale of ceritinib design was based on the reversal of piperidine and the inclusion of methyl group para to the isopropoxy moiety at the aniline ring, minimizing the possibility of forming reactive species, and consequently the unwanted properties of the compound 7, while preserving the desirable biological and pharmacological activities of its predecessor drug (Figure 1B).29
Figure 1.
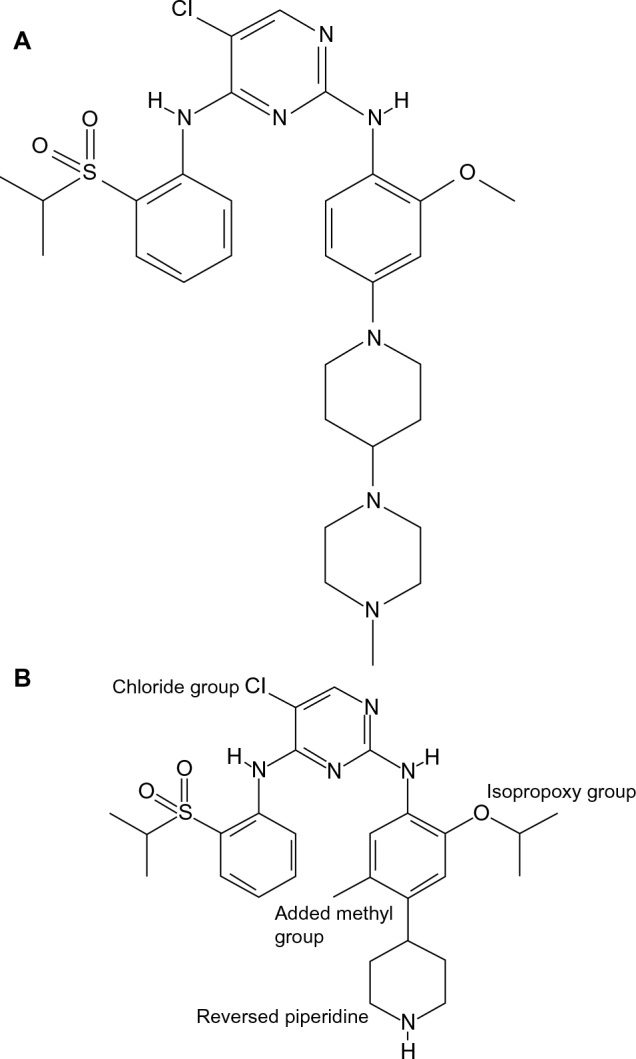
Chemical structure of TAE684 (A) and LDK378 (B).
Notes: This figure depicts the structural determinants of Ceritinib potency. The group isopropoxy at the aniline ring confers an improved kinase selectivity of Ceritinib, whereas the reversal of piperidine along with the methyl group para the isopropoxy are thought to minimize the possibility of reactive adducts formation.
Preclinical data
Ceritinib exhibits 20-fold greater enzymatic inhibitory potency than crizotinib in vitro. This can be explained by the chemical structural difference between the two drugs, essentially the chlorine in the fifth position of the pyrimidine ring enhances ceritinib’s interactions with the mutant residues at the gatekeeper position of the ALK kinase domain.25 In addition, molecular dynamic simulation analyses have shown an increased hydrophobic interaction between the isopropyl group of ceritinib and the mutated ALK, as well as the electrostatic interactions between the K1150 amino acid and sulfonyl group of the drug.30 When tested against a panel of lung cancer cell lines bearing ALK rearrangements, ceritinib had more potent activity than crizotinib in terms of ALK phosphorylation suppression and cell growth inhibition. However, ceritinib was not potent against NSCLC and breast cancer cell lines driven by other molecular alterations such as KRAS, EGFR, PI3K, or Her2, indicating a high selectivity of the drug for the EML4-ALK fusion protein.25 Additionally, in xenograft models using crizotinib-naïve H2228 ALK-rearranged cell lines, ceritinib demonstrated a more durable antitumor effect than crizotinib after drug discontinuation. In parallel, crizotinib-refractory H2228 xenografts were generated by treatment with incremental doses of crizotinib. Sequencing of tumors that progressed while on crizotinib detected G1202R, C1156Y, or I1171T mutations in 6 out of the 80 treated animals.25 Interestingly, ceritinib had marked antitumor activity against the wild-type EML4-ALK and the I1171T mutation, less activity against the C1156Y mutation, and showed no activity against the G1202R mutation.25 On the other hand, high antitumor activity was observed in vivo and in vitro by using ceritinib against ALK-rearranged cell lines derived from patients with acquired refractoriness to crizotinib and bearing the two most common EML4-ALK mutations: L1196M and G1269A.25
Clinical efficacy
Promising results for ceritinib in ALK-positive NSCLC were demonstrated in early phase clinical development. Interestingly, its pronounced antitumor activity was not only limited to crizotinib-naïve patients, but was also observed in crizotinib-refractory patients. In ASCEND-1, the first Phase I clinical trial testing ceritinib, 59 patients with advanced cancer carrying genetic alterations in ALK, received LDK378 with dose escalation from 50 to 750 mg once daily.9 The maximum tolerated dose was 750 mg once daily. This was followed by an expansion cohort in which 71 additional patients were treated with ceritinib at the maximum tolerated dose as determined in the first phase. Among 114 patients with NSCLC who received at least 400 mg of ceritinib/d, the overall response rate was 58%. The majority of patients with NSCLC treated with ceritinib had received crizotinib previously. Interestingly, in the subgroup of 80 patients with NSCLC who previously were treated with crizotinib, the response rate was 56%, suggesting that ceritinib may be effective for the treatment of patients with ALK-rearranged NSCLC who have developed crizotinib resistance. Nevertheless, among patients with NSCLC who received at least 400 mg of ceritinib/d, the median PFS was 7.0 months (95% CI: 5.6–9.5). Among the patients with NSCLC who had previously received crizotinib, the PFS was 6.9 months (95% CI: 5.3–8.8), whereas in patients with no prior exposure to crizotinib, the median PFS was higher at 10.4 months (4.6 to could not be estimated). Clinical activity to CNS metastases was also observed in certain patients. Among the 14 patients with brain metastases who were treated at the 750 mg dose, the overall intracranial response rate was 50%.31 Unlike crizotinib, ceritinib has an improved bioavailability to brain tissue and increased CNS penetration, which can explain why it has better CNS activity. More recently, a Japanese Phase I study has demonstrated a similar antitumor activity with alectinib in ALK-positive lung cancer irrespective of prior treatment with ALK inhibitor. In particular, two out of four patients previously treated with alectinib had partial response.32
On the basis of its profound clinical activity, ceritinib was approved in April 2014 by the FDA for the treatment of ALK-positive metastatic NSCLC following treatment with crizotinib. Two Phase III trials are currently recruiting ALK-positive metastatic NSCLC patients, one comparing ceritinib with chemotherapy (pemetrexed or docetaxel) after progression on both crizotinib and platinum-based chemotherapy (ASCEND-5), and the other comparing ceritinib with first-line chemotherapy (pemetrexed with platinum) in treatment-naïve patients (ASCEND-4).33,34 The results of these two major trials are eagerly awaited since it helps to better define the position of this drug as well as the optimal timing of initiation in order to achieve an improved clinical outcome.
Clinical safety and tolerability
Ceritinib is generally well tolerated. However, treating oncologists should be aware of a number of significant adverse events that may require dose adjustment or treatment discontinuation. The most common adverse events included diarrhea (86%), nausea (80%), vomiting (60%), abdominal pain (54%), fatigue (52%), decreased appetite (34%), and constipation (29%). The most common laboratory abnormalities are decreased hemoglobin (84%), increased alanine transaminase (80%), increased aspartate transaminase (75%), creatinine elevation (58%), hyperglycemia (49%), decreased phosphate (36%), and increased lipase (29%). Dose reduction was required in 74% of the patients, and drug discontinuation was needed in 10% of patients. Less common side effects include neuropathy, vision disturbances, prolonged QT intervals, and bradycardia. The most common adverse events that led to treatment discontinuation were pneumonia, interstitial lung disease (ILD)/pneumonitis, and anorexia. In particular, ILD/pneumonitis occurred in 4% of ceritinib-treated patients. Development of a cough or shortness of breath while on ceritinib therapy should alert clinicians to rule out ILD/pneumonitis. Ceritinib should be permanently discontinued in the instance ILD/pneumonitis of any grade being diagnosed. Fatal adverse events were reported in 5% of treated patients and resulted from pneumonia, respiratory failure, ILD/pneumonitis, pneumothorax, gastrointestinal (GI) bleeding, general health deterioration, tuberculosis, cardiac tamponade, and sepsis (Table 2).31,35
Table 2.
Most common adverse reactions associated with the usage of ceritinib
| All grades (%) | Grades 3–4 (%) | Dose reduction or discontinuation (%) | |
|---|---|---|---|
| Most common clinical adverse reaction | |||
| Diarrhea | 86 | 6 | 16 |
| Nausea | 80 | 4 | 20 |
| Vomiting | 60 | 4 | 16 |
| Abdominal pain | 54 | 2 | |
| Fatigue | 52 | 5 | |
| Decreased appetite | 34 | 1 | |
| Constipation | 29 | 0 | |
| Most common laboratory adverse reaction | |||
| Decreased Hg | 84 | 5 | |
| Increased ALT | 80 | 27 | 29 |
| Increased AST | 75 | 13 | 16 |
| Increased creatinine | 58 | 2 | |
| Hyperglycemia | 49 | 13 | |
| Hypophosphatemia | 36 | 7 | |
| Increased lipase | 29 | 10 | |
| Less common clinical and laboratory adverse reactions | |||
| Neuropathy | 17 | ||
| Vision disturbances | 9 | ||
| Prolonged QT interval | 4 | ||
| Bradycardia | 3 | ||
| ILD/pneumonitis | 4 | 3 | |
| Increased bilirubin | 15 | 1 | |
| Rash | 16 | 0 | |
Pharmacokinetics
Ceritinib is available as a 150 mg capsule. The recommended dose for ALK-positive NSCLC is 750 mg oral once daily. Oral ceritinib is absorbed, with peak plasma concentration after first dose achieved 6 hours postingestion. The steady-state drug level is reached after approximately 15 days of continuous daily oral administration. The absolute drug bioavailability has not been determined. Systemic exposure of ceritinib is increased when administered with meals and may result in increased toxicity. For this reason, the drug should be administered on an empty stomach at least 2 hours prior to or after a meal.35 Ceritinib is 97% bound to human plasma proteins. It is metabolized primarily by the liver CYP3A enzymes and the metabolites are excreted mainly in the feces.31 Thus, caution should be used when ceritinib is coadministered with other agents metabolized by this system. The mean drug half-life elimination is estimated at 41 hours. No dosage adjustment is necessary in the presence of preexisting kidney dysfunction or mild hepatic impairment. Monitoring parameters should include complete blood counts, renal and liver function tests, glycemic indices, and cardiac monitoring (essentially heart rate and QTc). Patients taking ceritinib should also be monitored for any sign or symptoms of toxicity (mainly pulmonary and GI). Detailed information on dose adjustment for toxicity can be found in the product insert package.35 Treatment should be discontinued if a patient is unable to tolerate a dose as low as 300 mg daily.
Pharmacodynamics
Ceritinib (LDK378) is a highly potent and selective ATP-competitive ALK tyrosine kinase inhibitor.29 The half-maximal inhibitory concentration (IC50) of ceritinib is approximately 150 pmol/L, which is 20-fold lower that than of crizotinib. In addition to ALK, ceritinib also inhibits other tyrosine kinase receptors including the IGF1 (insulin-like growth factor 1) and insulin receptors, and at higher concentrations, ROS1.36,37 Ceritinib may cause hyperglycemia likely as a result of insulin activity inhibition. Ceritinib has demonstrated activity in crizotinib-resistant tumor in xenografts models. Specifically, it showed efficacy against different ALK mutations including, L1196M, G1269A, I1171T, and S1206Y crizotinib-resistant mutations. However, ceritinib is not active against G1202R mutation.25 Interestingly, ceritinib has shown activity against ALK-positive lung cancer cells derived from patients who failed alectinib.38 Unlike crizotinib, ceritinib does not inhibit MET activity.9
Therapeutic challenges with ceritinib
Mechanisms of resistance to ceritinib
When tested against a panel of engineered cancer cells driven by one of the nine different crizotinib-resistance ALK mutations, potent antigrowth efficacy was observed with ceritinib in cells expressing L1196M, G1269A, S1206Y, and I1171T ALK mutations. C1156Y, G1202R, 1151T-ins, L1152R, and F1174C secondary ALK mutation conferred resistance to ceritinib. These data were confirmed in a xenograft model when ceritinib failed to overcome crizotinib resistance in the H2228 tumor line carrying the G1202R mutation.25 Biopsies of 11 tumors from patients with acquired resistance to ceritinib reported G1202R or F1174C/V ALK mutation in five patients. Of note, crystal structural analyses of interaction ALK/ceritinib demonstrated that the G1202R mutation is associated with reduced binding affinity to ALK due to steric obstruction. Further studies are needed to characterize mechanism of resistance for the F1174 mutation.25 In patient-derived models of acquired resistance to the tyrosine kinase inhibitor, MEK activation mutation and Src signaling were identified as other mechanisms that mediate ceritinib resistance.36,39 Blockage of Src, or MEK, in these cells lines, while not effective when used alone, is able to restore sensitivity to ceritinib. Obtaining a tissue biopsy at the time of progression would be instrumental in identifying further mechanisms of ceritinib resistance.
Rational approaches to optimize the clinical benefit of ceritinib
The challenge in future studies is in designing effective combination therapies that tackle the problem of ceritinib resistance. In fact, several strategies to maximize the activity and tolerability of ceritinib, or to overcome ceritinib resistance in ALK-positive NSCLC patients have been suggested, including the following:
Combination with hsp90 inhibitor
ALK is a client protein for heat shock protein 90 (hsp90), a chaperone molecule integral for protein stability and protection from degradation. Multiple preclinical data have shown induced degradation of ALK protein following treatment with an hsp90 inhibitor.39,40 Other data have shown a synergism between hsp90 inhibitors and crizotinib, providing a rationale for a combination approach in patients failing crizotinib as a single agent.20,27 On the basis of these data, multiple early-phase clinical trials are currently exploring different classes of hsp90 inhibitors with ALK inhibitors. We are awaiting the results of a Phase Ib trial assessing the safety, tolerability, pharmacokinetics, and antitumor activity of the combination of ceritinib and the hsp90 inhibitor AUY922 in ALK rearranged NSCLC that has progressed on an ALK inhibitor (NCT01772797).
Combination with pemetrexed
Retrospective observations have suggested that patients with ALK-rearranged lung cancer have significantly greater PFS on pemetrexed compared with patients lacking ALK rearrangement, EGFR, and KRAS mutations.41 This has led to clinical trials evaluating pemetrexed with or without crizotinib, in patients who had progressed on the latter (NCT02134912). Further studies are required to investigate the safety and efficacy of adding pemetrexed to ceritinib.
Combination with other targeted therapy
The combination of ceritinib with compounds targeting the escape survival pathways such as MEK (MEK inhibitors) or Src (such as dasatinib) has a strong scientific rationale and has been tested in vitro.37,42 Its application in patients who develop resistance to ceritinib warrants further validation in clinical trials. Biopsy at the time of progression may help in the clinical decision-making process, based on the specific resistance mechanism(s) identified.
Combination with immunotherapy
Checkpoint inhibitors have demonstrated promising activities in metastatic NSCLC, leading to the approval of nivolumab in squamous lung cancer progressing on platinum-based therapy. Interestingly, sensitivity to PD1/PDL1 (programmed death 1/programmed death ligand 1) checkpoint inhibitors seems to be lower in nonsmoking patients; a subset of patient known to have higher likelihood for EGFR mutation and ALK rearrangements. Additionally, the EGFR mutation was found to activate the PD1/PDL1 pathway in the tumor microenvironment, which may contribute to immune escape.37,43,44 Although the underlying interplay between targeted therapy and antitumor immune response is complex, these two factors may influence each other from different aspects. By inducing rapid tumor regression, and consequently antagonizing the tumor-related immunosuppression while enhancing the expression of death receptors at tumor cell surface, it can further potentiate the efficiency of immune system to attack cancer cells. Additionally, the release of large amounts of antigenic debris subsequent to cell apoptosis can boost antigen presentation by dendritic cells, further triggering the immune response.45 Moreover, targeted therapies also more have a complex modulator effect on the tumor microenvironment, leading to activation of tumor-specific T-cells. On the other hand, immunotherapy may consolidate the significant tumor response induced by targeted therapy.45
Taken together, these findings show that targeted therapies and immunotherapy may offer a possible treatment synergism when employed together. These combinations are currently being explored in clinical trials. One study combining ceritinib with nivolumab will soon be initiated. However, a major challenge resides in defining how these agents should be dosed – simultaneously or sequentially?
Low-dose ceritinib
Since ceritinib absorption increases with fatty meals, which implies an increased in non-GI toxicity,31 an ongoing study is currently evaluating the systemic exposure and safety of a lower dose of ceritinib taken with a meal compared with the standard dose of the drug taken while fasting (NCT02299505). In the absence of published data, the 750 mg daily taken in the fasted state remains the standard therapy.
Conclusion and future perspectives
Since the identification of the EML4-ALK driver mutation in 2007, multiple ALK inhibitors have been developed to target this molecular alteration. Crizotinib is the first-in-class ALK inhibitor to receive FDA approval for the treatment of ALK-positive lung cancer. Despite the initial response to crizotinib, acquired resistance to the drug inevitably leads to disease progression. Ceritinib is the first second-generation TKI approved for the treatment of ALK-positive NSCLC in patients intolerant or progressed on crizotinib. Despite the promising activity of ceritinib, several unmet needs are awaiting further research, including 1) defining the optimal sequence of ALK inhibitors administration, 2) expanding their application to other potential indications in lung cancer, such as in the adjuvant and neo-adjuvant settings, and in combination with radiation, 3) exploring the drug efficacy in other solid and hematologic malignancies carrying the ALK rearrangements, and 4) establishing the drug impact on the patient’s quality of life and well-being.
Since its FDA approval in 2014, several ongoing trials are investigating ceritinib role for other indications (https://clinicaltrials.gov/) (Table 3), including its use in treatment-naïve ALK positive NSCLC, ROS-1 mNSCLC, and other ALK-positive malignancies (thyroid, cholangiocarcinoma, pancreas, and hematologic malignancies). Another challenge arising from the availability of multiple ALK inhibitors remains in defining the optimal first-line choice and then the sequence for therapies after ALK-positive NSCLC progression. Presently, it is not known if first-line ceritinib use in ALK-positive NSCLC will be superior to crizotinib. The ALEX trial, an ongoing randomized controlled trial comparing alectinib versus crizotinib in treatment-naïve ALK + NSCLC, will address this issue.46 However, this trial’s design does not allow the crossover among the treatment arms, limiting the ability to examine the impact of treatment sequencing (crizotinib followed by alectinib on progression versus alectinib first line) on outcome. An emerging groundbreaking trial, the NCI ALK Master protocol, will evaluate different next-generation ALK inhibitors with crizotinib in the first-line setting. Unlike the ALEX trial, crossover at the time of progression will be allowed, which will enable exploration the optimal strategy for the initial selection and subsequent selection of ALK-directed therapy (crizotinib followed by a next-generation ALK inhibitor versus next-generation ALK inhibitor followed by crizotinib upon progression).37
Table 3.
Some of the major ongoing clinical trials investigating the efficacy of ceritinib against different malignancies
| Clinical trial identifier | Study design | Treatment setting | Intervention | Primary endpoint |
|---|---|---|---|---|
| NCT01828099 | Phase III RCT | Metastatic treatment naïve ALK + NSCLC | Ceritinib versus platinum/Pemetrexed | PFS |
| NCT01828112 | Phase III RCT | Metastatic previously treated (with chemo and crizotinib) ALK + NSCLC | Ceritinib versus Docetaxel or Pemetrexed | PFS |
| NCT02289144 | Phase II | Locally advanced or metastatic ALK + anaplastic thyroid cancer | Ceritinib | PFS |
| NCT02374489 | Phase II | ROS1 or ALK overexpressed advanced cholangiocarcinoma | Ceritinib | ORR |
| NCT01964157 | Phase II | Unresectable NSCLC carrying ROS1 rearrangement | Ceritinib | ORR |
| NCT02393625 | Phase I | Relapsed locally advanced or metastatic ALK + NSCLC | Ceritinib plus nivolumab | MTD(br)ORR |
| NCT02321501 | Phase I | Locally advanced or metastatic previously treated solid tumor with an expansion in ALK + NSCLC | Ceritinib plus everolimus | MTD |
| NCT01685138 | Phase II | ALK + crizotinib naïve metastatic NSCLC | Ceritinib | ORR |
| NCT02336451 | Phase II | ALK + NSCLC with CNS metastasis | Ceritinib | ORR |
| NCT02299505 | Phase I | ALK + metastatic NSCLC | Low dose ceritinib taken with low-fat meal versus standard dose ceritinib at fasting | PK |
| NCT02227940 | Phase I | Advanced solid tumor or locally advanced/metastatic pancreatic cancer | Ceritinib combined with gemcitabine-based chemotherapy | MTD |
| NCT02343679 | Phase II | ALK + refractory/relapsed hematological malignancies | Ceritinib | ORR |
Abbreviations: PFS, progression free survival; ORR, overall response rate; MTD, maximum tolerated dose; CNS, central nervous system; NSCLC, Non-small-cell lung carcinoma; ALK, anaplastic lymphoma kinase; PK, pharmacokinetic.
Acquired resistance has emerged as a major hurdle preventing ALK targeting agents from having a long-term positive impact on patients. Additional research looking into the molecular basis of drug resistance will hopefully lead to more effective therapeutic strategies to overcome ALK inhibitor resistance. The combination of ceritinib with cytotoxic chemotherapy, agents targeting escape signaling pathways, and/or immunotherapy are all potential alternatives worth investigating in order to achieve improved clinical outcomes for ALK-positive NSCLC.
Footnotes
Disclosure
The authors report no conflicts of interests in this work.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.NSCLC Meta-Analyses Collaborative Group Chemotherapy in addition to supportive care improves survival in advanced non-small cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J Clin Oncol. 2008;26(28):4617–4625. doi: 10.1200/JCO.2008.17.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ardizzoni A, Boni L, Tiseo M, et al. Cisplatin-versus carboplatin-based chemotherapy in first-line treatment of advanced non-small cell lung cancer: an individual patient data meta-analysis. J Natl Cancer Inst. 2007;99(11):847–857. doi: 10.1093/jnci/djk196. [DOI] [PubMed] [Google Scholar]
- 4.Delbaldo C, Michiels S, Syz N, et al. Benefits of adding a drug to a single-agent or a 2-agent chemotherapy regimen in advanced non-small cell lung cancer: a meta-analysis. JAMA. 2004;292(4):470–484. doi: 10.1001/jama.292.4.470. [DOI] [PubMed] [Google Scholar]
- 5.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 9.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 12.Choi YL, Takeuchu K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2010;16(22):5581–5590. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 14.Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–19897. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamoto I, Nakagawa K. Echinoderm microtubule-associated protein-like 4-anaplastinc lymphoma kinase-targeted therapy for advanced non-small cell lung cancer: molecular and clinical aspects. Cancer Sci. 2012;103(8):1391–1396. doi: 10.1111/j.1349-7006.2012.02327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46(10):1773–1780. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus che-motherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 18.National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. Version 5. Fort Washington PA: NCCN; 2015. [Google Scholar]
- 19.Massarelli E, Papadimitrakopoulou V. Ceritinib for the treatment of late-stage (metastatic) non-small cell lung cancer. Clin Cancer Res. 2015;21(4):670–674. doi: 10.1158/1078-0432.CCR-14-1291. [DOI] [PubMed] [Google Scholar]
- 20.Iacono D, Chiari R, Metro G, et al. Future options for ALK-positive non-small cell lung cancer. Lung Cancer. 2015;87:211–219. doi: 10.1016/j.lungcan.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Loong H, Mok K, Leung L, Mok T. Crizotinib in the management of advanced non-small cell lung cancer. Future Oncol. 2015;11(5):735–745. doi: 10.2217/fon.14.314. [DOI] [PubMed] [Google Scholar]
- 22.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YL, Soda M, Yamashita T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 25.Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shackelford RE, Vora M, Mayhall K, Cotelingam J. ALK-rearrangements and testing methods in non-small cell lung cancer: a review. Genes Cancer. 2014;5:1–14. doi: 10.18632/genesandcancer.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sang J, Acquaviva J, Friedland JC, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3:430–443. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galkin A, Melnick J, Kim S, et al. Identification of NVP-TAE684, a potent, selective, efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104(1):270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (LAK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,3-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56:5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 30.Ni Z, Zhang TC. Computationally unraveling how ceritinib overcomes drug-resistance mutations in ALK-rearranged lung cancer. J Mol Model. 2015;21:175. doi: 10.1007/s00894-015-2716-z. [DOI] [PubMed] [Google Scholar]
- 31.Khozin S, Blumenthal G, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res. 2015;21(11):2436–2439. doi: 10.1158/1078-0432.CCR-14-3157. [DOI] [PubMed] [Google Scholar]
- 32.Nisho M, Murakami H, Horiike A, et al. Phase I study of ceritinib (LDK378) in Japanese patients with advanced anaplastic lymphoma kinase rearranged non-small cell lung cancer or other tumors. J Thorac Oncol. 2015;10:1058–1066. doi: 10.1097/JTO.0000000000000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novartis Pharmaceuticals LDK378 versus chemotherapy in ALK rearranged (ALK positive) patients previously treated with chemotherapy (platinum doublet) and Crizotinib. [Accessed July 17, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT01828112. NLM Identifier: NCT01828112.
- 34.Novartis Pharmaceutical LDK378 versus chemotherapy in previously untreated patients with ALK rearranged non-small cell lung cancer. [Accessed July 17, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT01828099. NLM Identifier: NCT01828099.
- 35.Zycadia™ (Ceritinib) [product information] East Hanover, NJ: Novartis Pharmaceuticals; 2014. Available from: www.pharma.us-novartis.com. [Google Scholar]
- 36.Cooper M, Chim H, Chan H, Durand C. Ceritinib: a new tyrosine kinase inhibitor for non-small cell lung cancer. Ann Pharmacother. 2015;49(1):107–112. doi: 10.1177/1060028014553619. [DOI] [PubMed] [Google Scholar]
- 37.Katayama R, Lovly C, Shaw A. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227–2235. doi: 10.1158/1078-0432.CCR-14-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20(22):5686–5696. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat shock protein 90 inhibitor, in patients with molecularly defined non-small cell lung cancer. J Clinc Oncol. 2010;28:4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Socinski MA, Goldman J, El-Hariri I, et al. A multicenter phase 2 study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Caner Res. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camidge DR, Kono SA, Lu X, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6(4):774–780. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crystal AS, Shaw A, Sequist L, et al. Patient-derived models of acquired resistance can identify effective drug combination for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azuma K, Ota K, Kawahara A, et al. association of PD-L1 overexpression with activating EGFR mutations in surgically resected non-small cell lung cancer. Ann Oncol. 2014;25:1935–1940. doi: 10.1093/annonc/mdu242. [DOI] [PubMed] [Google Scholar]
- 45.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roche Hoffman-La. ALEX study: a randomized, phase iii study comparing alectinib with crizotinib in treatment-naive anaplastic lymphoma kinase (alk)-positive advanced non-small cell lung cancer (NSCLC) patients. [Accessed July 17, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT02075840. NLM Identifier: NCT02075840.