

Abstract
Giving rise to both bone and cartilage during development, bone marrow-derived mesenchymal stem cells (hMSC) have the unique capacity to generate all the cells and tissues of the osteochondral interface. Utilizing a scaffold-free hMSC system, biphasic osteochondral constructs are incorporated with 2 types of growth factor-releasing microparticles to enable spatially organized differentiation. Gelatin microspheres (GM) releasing TGF-β1 are combined with hMSCs to form the chondrogenic phase. The osteogenic phase contains hMSCs only, mineral-coated hydroxyapatite microparticles (MCM), or MCM loaded with BMP-2, cultured in medium with or without BMP-2. After 4 weeks, TGF-β1 release from GM within the cartilage phase promotes formation of a matrix rich with glycosaminoglycans (GAG) and type II collagen, and appears to have a local inhibitory effect on osteogenesis. In the osteogenic phase, type X collagen and osteopontin are produced in all conditions. However, calcification occurs on the outer edges of the chondrogenic phase in some constructs cultured in media containing BMP-2, and alkaline phosphatase levels are elevated, indicating that BMP-2 releasing MCM provides better control over region-specific differentiation. The production of complex, stem cell-derived osteochondral tissues via incorporated bioactive microparticles could enable earlier in vivo implantation, potentially improving patient outcomes in the treatment of osteochondral defects.
Keywords: cartilage, bone, BMP (bone morphogenetic protein), TGF (transforming growth factor), hydroxyapatite
Graphical Abstract
A stem cell-derived, biphasic osteochondral construct is engineered with TGF-β1-releasing hydrogel microspheres in the chondrogenic phase and BMP-2-releasing hydroxyapatite microparticles in the osteogenic phase. Results demonstrate, for the first time, that spatial control of osteochondral differentiation can be achieved in a scaffold-free cellular construct via the incorporation of bioactive microparticles.
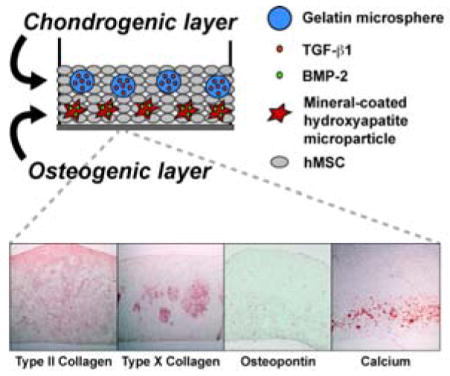
1. Introduction
The osteochondral interface consists of a layer of calcified cartilage between the deep zone of articular cartilage and the subchondral bone. Typically occurring as a result of traumatic injury or disease, osteochondral defects penetrate through this interfacial region, affecting both the surface cartilage and the underlying bone. Such defects are associated with reduced joint stability, and often lead to degenerative changes and osteoarthritis in the affected joint.[1] Current treatments for osteochondral defects include osteochondral autograft transfer system (OATS) or mosaicplasty, which involves the transfer of small cylindrical plugs of osteochondral tissue from a low-weight bearing region of the joint into the defect. However, the use of this treatment is limited by the amount of healthy cartilage available for grafting, and can cause donor site morbidity.[2] Additionally, though the OATS procedure initially results in a smooth articular surface, the grafted cartilage tissue typically does not integrate with the existing cartilage, leading to gaps between the host and grafted tissue and reducing the quality of repair.[3] Other surgical treatments include autologous chondrocyte implantation (ACI)[4] or marrow stimulation techniques such as microfracture and subchondral drilling,[5] but no single treatment to-date has been shown to effectively and consistently restore normal joint function.
As an alternative treatment strategy, tissue engineering therapies have been developed to promote repair and regeneration in patients with chondral (affecting the cartilage only) and osteochondral defects. In the case of joint damage affecting only the cartilage layer, osteochondral constructs could be anchored into the subchondral bone, potentially improving mechanical stability compared to a cartilage-only construct.[1] Although many advances have been made in the field of tissue engineering, complex tissues such as the osteochondral interface that contain multiple cell types with a distinct spatial organization present a unique challenge.[6]
Approaches to engineer osteochondral constructs have included layering chondrocytes and osteoblasts in various polymer scaffolds with defined structural or biochemical characteristics.[7] Though this approach has shown promising results, due to the low chondrocyte density in cartilage tissue and rapid dedifferentiation in monolayer culture, it may be difficult to obtain therapeutically relevant numbers of mature articular chondrocytes for these types of constructs.[8] Others have theorized that since bone and cartilage cells arise from a common progenitor, bone marrow-derived mesenchymal stem cells (MSCs), these cells could be used to generate all of the cells and tissues of the osteochondral interface.[9] Human MSCs (hMSCs) are a popular cell source for tissue engineering applications, as they are readily available in bone marrow, can be differentiated into multiple cell types of the mesenchymal lineage, and can be expanded for several passages in culture without losing their multipotency.[10] Several approaches have involved pre-differentiating MSCs down the chondrogenic and osteoblastic lineages in vitro prior to incorporation within a biomaterial scaffold, resulting in complex engineered osteochondral tissues derived from a single, abundant cell source.[11, 12] However, the extended culture times required to differentiate the MSCs prior to combining them within a single scaffold limits the clinical applicability of this type of construct.
Still another strategy involves seeding MSCs within a polymer scaffold that enables the spatial presentation of chondrogenic growth factors such as transforming growth factor-β1 (TGF-β1) or insulin-like growth factor-1 (IGF-1) in the cartilage region and osteogenic factors such as bone morphogenetic protein-2 (BMP-2) or BMP-4 in the bone region.[13, 14] This strategy circumvents difficulties with extended in vitro pre-culture times, as cell differentiation could occur in situ within the construct. With these types of systems, however, presence of the polymer matrix limits the concentration of cells that can be delivered to the defect, may interfere with the cell-cell interactions necessary for chondrogenesis, and could produce toxic degradation byproducts.[15] Further complicating these systems, the degradation rates of each layer of the polymer scaffold must be balanced with the rates of local tissue formation to preserve mechanical integrity.[16] Since bone-to-bone interfaces heal faster and more robustly than cartilage-to-cartilage interfaces,[9] the bone portion of the scaffold would be required to degrade more quickly than the cartilage portion. An inappropriate degradation rate of either phase of the polymer scaffold could compromise the repair process, as a scaffold degrading too rapidly could leave gaps in the healing tissue, or a scaffold degrading too slowly could inhibit tissue regeneration.[17] This could also present a problem with cell-free osteochondral implants, biomaterial-based constructs that function to deliver growth factors to the defect region and rely on the recruitment of cells from surrounding tissues following implantation.[13, 18–20] It may also not be possible to recruit a large enough number of endogenous cells to the defect space to elicit repair.
Cell-based “scaffold-free” approaches have also been proposed for engineering the osteochondral interface. Many of these approaches involve cellular constructs comprised of chondrocytes or stem cells in aggregates[21–23] or sheets,[24, 25] or consist of a cell-only cartilage layer atop a biomaterial scaffold that serves as the bone portion.[26–28] Though scaffold-free strategies avoid many of the issues with polymer-based scaffolds, stem cells alone may not receive appropriate signals in the correct amounts to differentiate into the complex tissues of the osteochondral interface,[9] and cellular aggregates may be difficult to localize within a defect. Partial scaffold-free approaches have similar problems to those of fully biomaterial-based scaffolds, with potential for material interference with healing or undesirable degradation by-products.
To address these issues, we designed an MSC-based osteochondral construct incorporating bioactive microparticles to enable spatial control of growth factor release and cell differentiation. Such an approach may circumvent problems with extended in vitro culture time, undesirable polymer matrix degradation rates, and mature cell sourcing.[29] The inclusion of growth factor-releasing microspheres within a densely cellular construct would enable the local delivery of factors to cells within the construct, avoiding problems with diffusion from the culture medium and uptake by cells at the periphery of the construct.[30–35] Importantly, incorporated bioactive microparticles have the potential to enable MSC differentiation after in vivo implantation, eliminating the time and expense associated with long-term in vitro culture in growth factor-containing media.
Here, we report the development of the first scaffold-free, stem cell-based biphasic osteochondral construct. This approach utilizes a single cell source (hMSCs) and a 2-step process to generate a complex, spatially organized tissue mimicking the structure and composition of the native osteochondral interface. Our hypothesis was that the spatially controlled delivery of chondrogenic and osteogenic factors from microparticles within a densely cellular hMSC construct could promote tissue-specific differentiation within each region of the construct. Incorporated gelatin microspheres (GM) enable rapid release of TGF-β1 to cells in the chondrogenic layer, and hydroxyapatite mineral-coated microparticles (MCM) facilitate the sustained delivery of BMP-2 to cells in the osteogenic layer. Furthermore, we hypothesized that the presence of BMP-2-loaded MCM would restrict the chondrocyte hypertrophy and osteogenic differentiation to the bone phase of the construct, reducing hypertrophic activity and osteogenesis in the cartilage phase by comparison to constructs cultured in medium containing BMP-2.
2. Results
2.1. Experimental Approach
To form the osteogenic layer of the osteochondral constructs, bone marrow-derived hMSCs were combined with MCM particles with or without loaded BMP-2 (as described in detail in the Experimental Section) and allowed to settle onto the membranes of Transwell inserts for 24 hours. Additional hMSCs were then combined with genipin-crosslinked gelatin microspheres loaded with TGF-β1 (as described in the Experimental Section) and gently added atop the osteogenic layer of all biphasic constructs to form the cartilage layer. Using this methodology, the diameter of the biphasic constructs was defined by the diameter of the Transwell inserts (12 mm). Constructs were formed using hMSCs from 2 donors, Donor A and Donor B. The specific conditions of osteochondral construct formation are presented in Table 1.
Table 1.
Experimental conditions of osteochondral construct formation.
| Condition | MCM | BMP-2 in Medium | BMP-2 in MCM |
|---|---|---|---|
| Exogenous (Exo.) BMP-2 | No | Yes | -- |
| MCM | Yes | No | No |
| MCM + BMP-2 | Yes | No | Yes |
| MCM + Exo. BMP-2 | Yes | Yes | No |
2.2. Microsphere Characterization
Gelatin microspheres were approximately spherical, with an average diameter of 48.4 ± 48.9 μm and a crosslinking level of 32.6 ± 6.1%. Characterization of TGF-β1 release from the formulation of GM and BMP-2 release from the formulation of MCM used here is reported in Dang et al. 2014. TGF-β1 was released from GM in collagenase-containing medium over a period of 10 days, with complete microsphere degradation and growth factor release occurring on day 10.[33] Roughly 60% of incorporated BMP-2 was released from MCM in PBS over a period of 60 days.[33]
2.3. Biochemical Analysis
After 4 weeks of culture, biphasic constructs from both donors were easily harvested by peeling them away the Transwell membranes. Similar biochemical trends were observed for both donors unless otherwise noted. DNA content was similar for all constructs containing MCM (Figure 1A). DNA content was lower in the Exogenous (Exo.) BMP-2 constructs than in all conditions containing MCM for each individual donor, but this difference was only significant for Donor A. The GAG content of all constructs exposed to BMP-2 was similar, but the MCM-only condition was lower for each individual donor (Figure 1B), with the difference being significant for Donor B versus the Exo. BMP-2 condition. The GAG/DNA in the Exo. BMP-2 condition was significantly higher than the other 3 conditions (Figure 1C) for Donor B, and significantly higher than MCM only for Donor A. The average ALP activity was higher in both conditions cultured in media containing BMP-2 than conditions without exogenous BMP-2, and the difference was significant for all conditions except MCM + Exo. BMP-2 in Donor A. Donor B was observed to have greater average ALP activity levels than Donor A when treated with exogenous BMP-2, and in the case of Donor B ALP levels for Exo. BMP-2 were significantly higher than MCM + Exo. BMP-2 (Figure 2A). Only minimal calcium was detected in the Exo. BMP-2 condition, but statistically equivalent amounts of calcium were present in the constructs incorporated with MCM for each donor, with higher average amounts for Donor B than Donor A (Figure 2B).
Figure 1.

DNA (A), GAG (B), and GAG per DNA (C) in osteochondral constructs after 4 weeks of culture. *p<.05 vs. MCM; **p<.05 vs. Exo. BMP-2; ***p<.05 vs. all other conditions.
Figure 2.

Alkaline phosphatase activity (A), and calcium content (B) in 4-week osteochondral constructs. **p<.05 vs. Exo. BMP-2; ***p<.05 vs. all other conditions; x p<.05 vs. MCM + Exo. BMP-2.
2.4. Safranin-O/Fast Green and Alizarin Red Histology
Histological sections were stained with Safranin-O to highlight the presence and distribution of GAG within the biphasic tissues. Representative sections from Donor A are shown (Figure 3). The “bottom” layer of the tissue was defined as the side nearest to the Transwell membrane where MCM (or cells only) were initially incorporated, and the “top” layer of the tissue was defined as the side facing away from the Transwell membrane, where GM containing TGF-β1 were incorporated. All GM appeared to be completely degraded at 4 weeks, and the regions where GM had initially been present were completely filled with cells and dense GAG-containing matrix. Constructs from the Exo. BMP-2 condition (without incorporated MCM) were uniformly stained with Safranin-O throughout (Figure 3A). All conditions containing MCM (MCM, MCM + BMP-2, and MCM + Exo. BMP-2) displayed an intense, uniform orange staining throughout the top ~2/3 of the tissues (Figure 3F, K, P). Regions within the bottom ~1/3 of the tissues, where the MCM were incorporated, stained less intensely for GAG and were more cellular. This was particularly apparent in the MCM + Exo. BMP-2 group (Figure 3P). Calcium staining indicated the presence of MCM in the lower region of all constructs from the MCM, MCM + BMP-2, and MCM + Exo. BMP-2 conditions (Figure 3J, O, T). In conditions containing both MCM and BMP-2, the region of red calcium staining was thicker and more distributed than in the MCM only condition (Figure 3O, T). Some calcification was also seen on the top side of the tissue in at least 1 sample from conditions cultured in media containing BMP-2 of each donor (Figure 3E).
Figure 3.

Cross-sections from osteochondral constructs stained for GAG (orange; A, F, K, P), type II collagen (red; B, G, L, Q), type X collagen (red; C, H, M, R), osteopontin (red; D, I, N, S) and calcium (red; E, J, O, T). Arrows indicate cell and matrix-filled regions where microspheres degraded. Scale bar = 100 μm for all images.
2.5. Immunohistochemistry for Type II Collagen, Type X Collagen, and Osteopontin
Generally, similar trends were observed in the distribution of IHC staining for both Donor B and Donor A. However, samples from Donor A stained more intensely for both types of collagen than Donor B, across all 4 conditions. Type II collagen was observed throughout constructs from all conditions, and stained more intensely towards the top edge of most constructs (Figure 3B, G, L, Q). Regions of less intense type II collagen stain filled with cells and matrix were observed where GM appear to have been initially present (black arrows). Type X collagen staining was noted in the central ~1/3 of constructs from all groups, but was much fainter in the MCM-only condition (Figure 3C, H, M, R). Osteopontin staining was localized to the bottom ~1/2 of the constructs, with the most intense staining observed along the bottom edge (Figure 3D, I, N, S). The region of osteopontin staining appeared thinner in the MCM only condition (Figure 3I).
2.6. Quantitative Image Analysis of Type X Collagen and Calcium Staining
Image analysis was performed on constructs stained for type X collagen or calcium to quantify the spatial distribution of these extracellular matrix components throughout the constructs. Constructs were divided into equal thirds by thickness, and the top, central, and bottom regions were isolated. In agreement with the gross visual findings, type X collagen was primarily located within the central 1/3 of the constructs (Table 2). The position and thickness of the regions of calcium staining in constructs from the MCM, MCM + BMP-2, and MCM + Exo. BMP-2 was also in agreement with the gross visual evaluation (Table 2). The MCM + BMP-2 constructs contained the thickest region of calcium staining, and the calcium-stained regions in both conditions containing MCM and treated with BMP-2 were significantly thicker than in the MCM only constructs. Osteochondral constructs from all conditions containing Exo. BMP-2 were significantly thicker than those containing MCM only.
Table 2.
Results of quantitative image analysis of type X collagen and calcium staining.
| Condition | Construct thickness (mm) | Type X collagen staining by region (%) | Calcium staining by region (%) | Thickness of calcium layer (mm) | ||||
|---|---|---|---|---|---|---|---|---|
| Top | Central | Bottom | Top | Central | Bottom | |||
| Exo. BMP-2 | 1.11 ± .05 | 9.4 ± 10.9 | 76.2 ± 3.4* | 14.4 ± 12.9 | 66.7 ± 57.7 | 0.0 ± 0.0 | 33.3 ± 57.7 | N/A |
| MCM | 0.95 ± .11** | 3.7 ± 4.3 | 75.1 ± 10.6* | 21.2 ± 10.6 | 1.4 ± 2.4 | 0.0 ± 0.0 | 98.6 ± 2.4* | 0.11 ± .01*** |
| MCM + BMP-2 | 1.10 ± .07 | 6.4 ± 9.4 | 88.5 ± 9.3* | 5.0 ± 7.4 | 0.0 ± 0.0 | 2.6 ± 4.4 | 97.4 ± 4.4* | 0.40 ± .02*** |
| MCM + Exo. BMP-2 | 1.14 ± .04 | 2.8 ± 2.3 | 80.2 ± 4.6* | 17.0 ± 6.5* | 0.0 ± 0.0 | 0.0 ± 0.0 | 100.0 ± 0.0* | 0.27 ± .03*** |
p<0.05 vs. other regions,
p<0.05 vs. Exo. BMP-2 and MCM + Exo. BMP-2,
p<0.05 vs. other conditions)
3. Discussion
Recent advances in the production of high-density stem cell constructs have demonstrated the utility of incorporating growth factor-releasing microparticles within the cellular constructs themselves, allowing the formation of bone and cartilage without culture in growth factor containing media.[30–35] TGF-β1-releasing polymer microspheres, when incorporated into high-density stem cell aggregates or sheets, can improve mechanical properties, enhance GAG-containing matrix production, and enable spatiotemporal control of neocartilage formation.[31, 32] Although promising chondrogenesis is achieved in these systems, differentiation into a single, cartilaginous phenotype may limit their applicability in the treatment of osteochondral defects penetrating through the subchondral bone. As the treatment of such defects may require both bone and cartilage templates, a more complex system that induces differentiation into both tissue types could be advantageous. Bone tissue engineering via endochondral ossification has been explored utilizing bioactive microparticle-incorporated high-density stem cell constructs, creating injectable or implantable systems to treat bone defects without requiring extended in vitro culture.[33, 34] In hMSC aggregates, BMP-2-loaded MCM alone[34] or in combination with TGF-β1-releasing GM[33] was used to successfully regulate both osteogenic and chondrogenic differentiation, though distinct spatial control of bone and cartilage formation was not achieved in these systems. The formation of a spatially organized, complex osteochondral tissue based on the self-assembly of stem cells had not yet been demonstrated.
In this study, we hypothesized that spatial control of chondrogenic and osteogenic differentiation within biphasic hMSC constructs could be attained via the spatially organized incorporation of TGF-β1-loaded gelatin microspheres and BMP-2-loaded mineral-coated hydroxyapatite microparticles. Genipin-crosslinked GM were utilized within the chondrogenic layer of the biphasic constructs due to their ability to release TGF-β1 in a controlled manner at rates dependent on the level of crosslinking[31, 32]. Within the hMSC constructs, cell-mediated proteolysis of the GM occurs, enabling growth factor release from the polymer matrix. This formulation of GM has been shown to release 100% of incorporated TGF-β1 over a period of 10 days in collagenase-containing medium,[33] but only a small fraction of the incorporated growth factor is released in medium without collagenase,[31] demonstrating the role of proteolytic degradation in releasing growth factor from within the charged hydrogel matrix. Based on our previous work, degradation of the GM within the high cell density constructs was expected to occur within 2 to 3 weeks.[31, 32] In agreement with those findings, complete microsphere degradation was observed histologically within the biphasic osteochondral constructs after 4 weeks.
Within the osteogenic layer of the hMSC constructs, MCM were used to provide a sustained delivery of BMP-2. HA was uniquely suited for this application as it is an osteoinductive material[36] deposited in the extracellular matrix (ECM) surrounding late hypertrophic chondrocytes during endochondral ossification.[37, 38] Furthermore, HA particles have a high protein-binding affinity, and can be modified with a mineral coating to provide tailorable growth factor release depending on the specific composition, dissolution rate and morphological properties of the mineralized surface.[39] The formulation of MCM utilized within this system has been shown to release approximately 60% of bound BMP-2 over a period of 60 days.[33] Within the biphasic osteochondral hMSC constructs, early delivery of TGF-β1 could promote chondrocyte differentiation, with a more prolonged delivery of BMP-2 to enable chondrocyte hypertrophy and calcification in the osteogenic region. This temporal sequence of tissue formation mimics the progression of endochondral ossification, the process by which hyaline cartilage is converted into bone.[40]
As was noted in a study with osteogenic MCM-incorporated hMSC aggregates, the inclusion of MCM within the biphasic constructs appeared to have a positive effect on the DNA content, potentially indicating improved cell viability.[34] The average GAG content was lower for the MCM-only condition than in all other constructs, though this difference was only significant for Donor B versus Exo. BMP-2. As BMP-2 has been shown to enhance GAG production by MSCs when administered in combination with TGF-β1,[41] it is reasonable that the GAG content was slightly improved with the addition of BMP-2 in particles or medium. The finding of higher GAG normalized to DNA in the Exo. BMP-2 constructs from Donor B indicates that the MCM may have reduced local GAG production in this system, as has also been demonstrated in hMSC aggregate cultures.[33] This is in agreement with the histological findings of uniform GAG staining throughout the Exo. BMP-2 condition, while MCM-containing constructs exhibit less intense GAG staining in the bottom 1/3 of the construct, on the osteogenic side (Figure 3P).
One of the goals of this study was to demonstrate that local release of BMP-2 from MCM incorporated within the cellular constructs could restrict the hypertrophy and calcification to the bone-like region of the biphasic constructs, reducing the amount of osteogenic differentiation in the cartilaginous phase compared to constructs cultured in medium containing BMP-2. The presence of exogenous BMP-2 may have led to the small areas of calcification observed histologically on both sides of some samples from the Exo. BMP-2 and MCM + Exo. BMP-2 conditions. ALP activity, an indicator of chondrocyte hypertrophy and early osteogenic differentiation,[42] was also higher in both conditions cultured in medium containing BMP-2. This could be due to the exposure of cells on all sides of those constructs to osteogenic growth factor, whereas the delivery of BMP-2 was restricted to the osteogenic phase in the MCM + BMP-2 condition.
Biochemical analysis revealed minimal amounts of calcium within the Exo. BMP-2 condition, indicating that the overall osteogenic differentiation of these constructs from both donors was limited at 4 weeks. The local delivery of TGF-β1 from degrading GM in the first 2–3 weeks may have delayed osteogenesis within this system, as other studies have shown that the sustained release of TGF-β may have an inhibitory effect on ALP activity and mineral deposition in hMSCs.[43] The calcium contents of the MCM-incorporated conditions were similar, regardless of the presence of BMP-2. A portion of the quantified calcium in the MCM-containing constructs was likely due to the calcium in the particles themselves, which were visualized histologically within the bottom phase of the constructs (Figure 3J, O, T).
Generally, constructs from Donor A stained more intensely for both types of collagen than those from Donor B, indicating donor-to-donor variability as has been previously observed.[32] Type II collagen, a major component of articular cartilage, was detected throughout constructs from all conditions, with the most intense staining present towards the top of many of the constructs, likely due to the local delivery of TGF-β1 from GM. The less-intense staining observed in cell- and matrix-filled regions where GM had initially been present could indicate a less-mature cartilaginous ECM in those areas, where the matrix had been remodeled as the gelatin degraded within the first 2 to 3 weeks of culture. These regions of remodeled tissue stained positively for Safranin-O, likely because GAG begins to accumulate within chondrogenic hMSC cultures before type II collagen is detected.[42] Type X collagen, a marker of chondrocyte hypertrophy, was observed in the central areas of constructs from all conditions but was notably fainter in the MCM-only constructs, probably due to the pro-hypertrophic effects of BMP-2 in the other 3 conditions.[44] Osteopontin (OP), a marker of both osteogenesis and chondrocyte hypertrophy,[45] was visualized in the bottom phase of all constructs, but was also reduced in constructs without BMP-2 (Fig. 3I). The absence of staining for type X collagen and OP in the upper regions of biphasic constructs in the Exo. BMP-2 condition may indicate that localized delivery of TGF-β1 had an inhibitory effect on hypertrophy and osteogenesis in the upper layer, allowing formation of an oriented osteochondral tissue even in media containing containing BMP-2.
Derived from MSCs and TGF-β1-releasing GM, the upper layer of the osteochondral constructs formed neocartilage containing cells within a dense ECM rich in type II collagen and GAGs, similar to native articular cartilage. The central and lower portions of the constructs seem to mimic the composition of deep zone articular cartilage, the layer nearest the articular cartilage-bone interface. Directly above the calcified tidemark, native deep zone cartilage contains GAG, type II collagen, type X collagen, and osteopontin.[38] The mineralized region on the bottom portion of the constructs is analogous to the tidemark of calcified cartilage, which forms slightly below hypertrophic chondrocytes expressing type X collagen at the base of mature articular cartilage during endochondral ossification.[38] Though these tissues are compositionally similar to the native osteochondral interface, other factors such as collagen fiber orientation play a critical role in the functional properties of the osteochondral interface. Future studies will explore the role of, for example, mechanical stimulation of the engineered constructs, as this has been shown to have a critical role in the zonal organization of osteochondral tissues.[46]
4. Conclusion
These findings demonstrate, for the first time, that incorporated bioactive microparticles can be used to direct complex, region-specific differentiation within high-density stem cell-derived tissues. Spatial control of cellular differentiation was attained in biphasic osteochondral constructs via incorporation of gelatin microspheres releasing TGF-β1 in the chondrogenic layer and mineral-coated hydroxyapatite microparticles releasing BMP-2 in the osteogenic layer. The ability to form osteochondral tissues without necessitating a polymer scaffold or extended culture in growth factor-containing media could increase the clinical utility of this type of treatment. Additionally, due to the modular nature of this system, incorporated bioactive microparticles could be used to produce other complex, spatially-organized tissues, depending on the type(s) of cells, microparticles, and bioactive factors used.
Experimental Section
hMSC Isolation and Culture
Bone marrow aspirates were harvested from 2 adult donors with informed, signed consent under a protocol approved by the University Hospitals of Cleveland Institutional Review Board. Mononuclear cells were isolated and plated at a concentration of 1.8x105 cells cm−2 in growth medium consisting of low glucose Dulbecco’s modified Eagle’s medium (DMEM-LG; Sigma) with 10% pre-selected fetal bovine serum (FBS; Gibco)[47] and penicillin/streptomycin (1%) as previously described.[48] The culture medium was changed to remove nonadherent cells after 2–3 days, and growth medium supplemented with FGF-2 (10 ng mL−1) was added and changed every 3 days thereafter.[49] Cells were used at passage 3.
Gelatin Microsphere Synthesis and TGF-β1 Loading
Gelatin microspheres were produced using acidic gelatin (11.1 wt.%; Sigma) according to an established protocol[50] with modifications as previously described.[31–33] Microspheres were crosslinked in genipin (1 wt.%; Wako USA) at room temperature for 2.5 hours, then collected by filtration, washed, and lyophilized. Crosslinked microspheres were UV sterilized for 10 minutes prior to use. Growth factor loading was accomplished by adding a small volume of phosphate buffered saline (PBS, pH 7.4) containing TGF-β1 (Peprotech) to sterile microspheres at a concentration of 400 ng TGF-β1 per mg microspheres and incubating for 2 hours at 37°C. The volume of growth factor-containing solution added was much less than the equilibrium swelling volume of the microspheres, ensuring complete absorption.[51]
Crosslinked GM were imaged under light microscopy and measured using Image J software (N=472). The degree of crosslinking was determined by ninhydrin assay as previously described.[52] The concentration of free amines was determined by comparison to glycine standards, with degree of crosslinking defined as the percentage of free amines that were reacted with the crosslinking agent.
Mineral-Coated Hydroxyapatite Microparticle Synthesis and BMP-2 Loading
Hydroxyapatite (HA) microparticles (3–5 μm diameter; Plasma Biotal LTD) were mineral- coated by incubation in modified simulated body fluid containing HCO3− (4.2 mM; pH 6.8) for 1 week as previously described.[33, 39] The resultant MCM were UV sterilized for 10 minutes prior to use. Growth factor loading was accomplished by adding PBS containing BMP-2 (R&D Systems) to sterile MCM to a final concentration of 6400 ng BMP-2 per mg MCM as previously described.[33] For MCM without BMP-2, particles were incubated similarly with PBS only.
Production of Biphasic Microsphere-Incorporated hMSC Constructs
To form the osteogenic layer, 12 mm Transwell inserts (3 μm pore size; Corning) were incubated with 1 mL of growth medium without FGF-2 for approximately 1 hour prior to use. hMSCs were combined with MCM particles (with or without BMP-2) in defined osteogenic medium [DMEM-HG with ITS+ Premix (1%; BD Biosciences), ascorbate-2-phosphate (50 μg mL−1; Wako USA), dexamethasone (10−7 M; MP Biomedicals), nonessential amino acids (1%; HyClone), sodium pyruvate (1%; HyClone), penicillin/streptomycin (1%; Gibco), and β-glycerophosphate (5 mM)] with or without exogenous BMP-2 at a concentration of 4x106 cells and 0.8 mg MCM per mL. 1 mL of osteogenic medium was added to the well plates outside the Transwells, and 500 μL of the hMSC/MCM suspension was added inside the Transwells and allowed to settle onto the membranes for 24 hours.
To form the chondrogenic layer, hMSCs were combined with TGF-β1-loaded gelatin microspheres and suspended in chemically defined chondrogenic medium [DMEM-HG with ITS+ Premix (1%; BD Biosciences), ascorbate-2-phosphate (37.5 μg mL−1; Wako USA), dexamethasone (10−7 M; MP Biomedicals), nonessential amino acids (1%; HyClone), sodium pyruvate (1%; HyClone), and penicillin/streptomycin (1%; Gibco)] at a concentration of 4x106 cells and 3 mg microspheres per mL. 24 hours after formation of the osteogenic layer, medium was carefully aspirated from the outside and inside of the Transwells, and 2 mL fresh osteogenic medium was added outside the Transwells. Inside each Transwell, 500 μL of the cell and gelatin microsphere suspension was gently added and allowed to settle atop the osteogenic layer. The medium was replaced with 2.5 mL of fresh osteogenic medium (with or without 100 ng mL−1 BMP-2) 24 hours later and every other day thereafter for 4 weeks of culture. The specific conditions of construct formation are described in Table 1.
Biochemical Analysis
Biphasic tissues (N=4) were harvested for analysis after 4 weeks of culture. Three 3-mm diameter punches were taken from each tissue for biochemical analysis and thickness measurements. Remaining portions were processed for histology and immunohistochemistry. Tissue punches designated for biochemical analysis (N≥3) were measured with calipers to determine thickness, and then digested and assayed for DNA, glycosaminoglycan (GAG), alkaline phosphatase (ALP), and calcium as previously described.[33] Briefly, tissue punches were homogenized on ice for 60 s in papain buffer (1 mL) containing papain (25 μg mL−1; Sigma), L-cysteine (2 mM; Sigma), sodium phosphate (50 mM), and EDTA, (2 mM, pH 6.5). Homogenate (0.5 mL) was combined with ALP lysis buffer [MgCl2 (1 mM), ZnCl2, (20 μM) and octyl-beta-glucopyranoside (0.1%) in tris buffer (10 mM, pH 7.4)]. Following a 30-minute incubation with p-nitrophenol phosphate at 37°C, ALP activity was determined by the amount of 4-nitrophenol present in the samples. The other 0.5 mL of homogenate was papain-digested overnight at 65°C. The following day, a portion of the sample was treated with HCl (1 M) to dissolve the MCM and assayed for calcium content using o-Cresophtalein complexone dye. The remainder of the sample was treated with EDTA (10%) in Tris-HCl (0.05 M), and DNA and GAG were quantified by Quant-iT PicoGreen dsDNA assay (Invitrogen) and dimethylmethylene blue assay,[53] respectively.
Histological and Immunohistochemical (IHC) Analysis
Tissue portions designated for histology and IHC (N=4) were fixed overnight in neutral buffered formalin and paraffin-embedded, and 5 μm sections were sliced. Sections were stained for GAG content via Safranin-O/Fast Green and for calcium via Alizarin Red S. The presence of collagen types II and X and osteopontin was assessed by IHC as previously described. [33] Briefly, sections were deparaffinized and rehydrated, and epitope retrieval was performed with pronase (for type II and X collagen staining) or citrate buffer (for osteopontin staining). Primary antibodies for collagen type II (II-II6B3; Developmental Studies Hybridoma bank), collagen type X (ab49945; Abcam), and osteopontin (ab8448; Abcam) were used along with the Histostain-Plus Bulk kit (Invitrogen) according to the manufacturer’s instructions. Aminoethyl carbazole (AEC; Invitrogen), a red chromogen, was utilized to visualize the antibody staining. Slides were mounted with glycerol vinyl alcohol (Invitrogen) and imaged using an Olympus BX61VS microscope (Olympus).
Quantitative Image Analysis
Image analysis was performed using Image J version 1.47t software to obtain quantitative measurements of the spatial distribution of type X collagen and alizarin red staining in histological sections of constructs from Donor A. To evaluate the spatial distribution of staining, constructs were divided into equal thirds by thickness (top, central, and bottom regions), and the fraction of total staining in each region was quantified. The thickness of the layer of calcium staining in the bottom region was also quantified via image analysis of the histological sections. For both the spatial distribution and calcium layer thickness analyses, measurements were averaged from images of histological sections from N 3 constructs per condition.
Statistical Analysis
For the biochemical analysis and thickness measurements, comparisons were made among the 4 conditions for each donor. For the image analysis of spatial distribution of staining, comparisons were made between the 3 regions (top, central, and bottom) for each condition. All values are reported as mean ± standard deviation. One-way ANOVA with Tukey's post hoc tests was performed using GraphPad InStat 3.06 software, with values of p<0.05 considered statistically significant.
Acknowledgments
The authors would like to thank Amad Awadallah for technical assistance. The authors gratefully acknowledge funding from the National Institutes of Health (R01AR063194; T32AR007505), and the AO Foundation.
Contributor Information
Dr. Loran D. Solorio, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
Lauren M. Phillips, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
Alexandra McMillan, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA.
Dr. Christina W. Cheng, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
Phuong N. Dang, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
Julia E. Samorezov, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
Dr. Xiaohua Yu, Departments of Biomedical Engineering and Orthopedics and Rehabilitation, University of Wisconsin, Madison, WI, 53706, USA
Prof. William L. Murphy, Departments of Biomedical Engineering and Orthopedics and Rehabilitation, University of Wisconsin, Madison, WI, 53706, USA, AO Foundation Collaborative Research Center, Clavadelerstrasse 8, Davos, 7270, Switzerland
Prof. Eben Alsberg, Email: eben.alsberg@case.edu, Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA, AO Foundation Collaborative Research Center, Clavadelerstrasse 8, Davos, 7270, Switzerland, Department of Orthopaedic Surgery, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH, 44106, USA
References
- 1.Martin I, Miot S, Barbero A, Jakob M, Wendt D. J Biomech. 2007;40(4):750. doi: 10.1016/j.jbiomech.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Filardo G, Kon E, Perdisa F, Balboni F, Marcacci M. Int Orthop. 2014 doi: 10.1007/s00264-014-2322-1. [DOI] [PubMed] [Google Scholar]
- 3.Raub CB, Hsu SC, Chan EF, Shirazi R, Chen AC, Chnari E, Semler EJ, Sah RL. Osteoarthritis Cartilage. 2013;21(6):860. doi: 10.1016/j.joca.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biant LC, Bentley G, Vijayan S, Skinner JA, Carrington RW. Am J Sports Med. 2014 doi: 10.1177/0363546514539345. [DOI] [PubMed] [Google Scholar]
- 5.Steinwachs MR, Guggi T, Kreuz PC. Injury. 2008;39(Suppl 1):S26. doi: 10.1016/j.injury.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Chen H, Li P, Diao H, Zhu S, Dong L, Wang R, Guo T, Zhao J, Zhang J. Biomaterials. 2011;32(21):4793. doi: 10.1016/j.biomaterials.2011.03.041. [DOI] [PubMed] [Google Scholar]
- 7.Giannoni P, Lazzarini E, Ceseracciu L, Barone AC, Quarto R, Scaglione S. J Tissue Eng Regen Med. 2012 doi: 10.1002/term.1651. [DOI] [PubMed] [Google Scholar]
- 8.Darling EM, Athanasiou KA. J Orthop Res. 2005;23(2):425. doi: 10.1016/j.orthres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Caplan AI, Elyaderani M, Mochizuki Y, Wakitani S, Goldberg VM. Clin Orthop Relat Res. 1997;(342):254. [PubMed] [Google Scholar]
- 10.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Science. 1999;284(5411):143. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 11.Lam J, Lu S, Lee EJ, Trachtenberg JE, Meretoja VV, Dahlin RL, van den Beucken JJ, Tabata Y, Wong ME, Jansen JA, Mikos AG, Kasper FK. Osteoarthritis Cartilage. 2014 doi: 10.1016/j.joca.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng HW, Luk KD, Cheung KM, Chan BP. Biomaterials. 2011;32(6):1526. doi: 10.1016/j.biomaterials.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Re'em T, Witte F, Willbold E, Ruvinov E, Cohen S. Acta Biomater. 2012;8(9):3283. doi: 10.1016/j.actbio.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 14.Dormer NH, Singh M, Wang L, Berkland CJ, Detamore MS. Ann Biomed Eng. 2010;38(6):2167. doi: 10.1007/s10439-010-0028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi Y, Du Y, Li W, Dai X, Zhao T, Yan W. Knee Surg Sports Traumatol Arthrosc. 2014;22(6):1424. doi: 10.1007/s00167-012-2256-3. [DOI] [PubMed] [Google Scholar]
- 16.Kretlow JD, Klouda L, Mikos AG. Adv Drug Deliv Rev. 2007;59(4–5):263. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Bichara DA, Bodugoz-Sentruk H, Ling D, Malchau E, Bragdon CR, Muratoglu OK. Biomed Mater. 2014;9(4):045012. doi: 10.1088/1748-6041/9/4/045012. [DOI] [PubMed] [Google Scholar]
- 18.Lu S, Lam J, Trachtenberg JE, Lee EJ, Seyednejad H, van den Beucken JJ, Tabata Y, Wong ME, Jansen JA, Mikos AG, Kasper FK. Biomaterials. 2014 doi: 10.1016/j.biomaterials.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyes R, Delgado A, Solis R, Sanchez E, Hernandez A, San Roman J, Evora C. J Biomed Mater Res A. 2014;102(4):1110. doi: 10.1002/jbma.34769. [DOI] [PubMed] [Google Scholar]
- 20.Kim K, Lam J, Lu S, Spicer PP, Lueckgen A, Tabata Y, Wong ME, Jansen JA, Mikos AG, Kasper FK. J Control Release. 2013;168(2):166. doi: 10.1016/j.jconrel.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JI, Sato M, Kim HW, Mochida J. Eur Cell Mater. 2011;22:275. doi: 10.22203/ecm.v022a21. [DOI] [PubMed] [Google Scholar]
- 22.Cheuk YC, Wong MW, Lee KM, Fu SC. J Orthop Res. 2011;29(9):1343. doi: 10.1002/jor.21339. [DOI] [PubMed] [Google Scholar]
- 23.Yoshioka T, Mishima H, Kaul Z, Ohyabu Y, Sakai S, Ochiai N, Kaul SC, Wadhwa R, Uemura T. J Tissue Eng Regen Med. 2011;5(6):437. doi: 10.1002/term.329. [DOI] [PubMed] [Google Scholar]
- 24.Oda K, Mori K, Imai S, Uenaka K, Matsusue Y. J Orthop Sci. 2014;19(4):637. doi: 10.1007/s00776-014-0574-7. [DOI] [PubMed] [Google Scholar]
- 25.Mainil-Varlet P, Rieser F, Grogan S, Mueller W, Saager C, Jakob RP. Osteoarthritis Cartilage. 2001;9(Suppl A):S6. doi: 10.1053/joca.2001.0438. [DOI] [PubMed] [Google Scholar]
- 26.Shimomura K, Moriguchi Y, Ando W, Nansai R, Fujie H, Hart DA, Gobbi A, Kita K, Horibe S, Shino K, Yoshikawa H, Nakamura N. Tissue Eng. Part A. 2014 doi: 10.1089/ten.tea.2013.0414. [DOI] [PubMed] [Google Scholar]
- 27.Miyagi S, Tensho K, Wakitani S, Takagi M. J Orthop Sci. 2013;18(3):471. doi: 10.1007/s00776-013-0368-3. [DOI] [PubMed] [Google Scholar]
- 28.Lee WD, Hurtig MB, Kandel RA, Stanford WL. Tissue Eng Part C Methods. 2011;17(9):939. doi: 10.1089/ten.TEC.2011.0147. [DOI] [PubMed] [Google Scholar]
- 29.Solorio LD, Vieregge EL, Dhami CD, Alsberg E. Tissue Eng Part B Rev. 2013;19(3):209. doi: 10.1089/ten.teb.2012.0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solorio LD, Fu AS, Hernandez-Irizarry R, Alsberg E. J Biomed Mater Res A. 2010;92(3):1139. doi: 10.1002/jbm.a.32440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solorio LD, Vieregge EL, Dhami CD, Dang PN, Alsberg E. J Control Release. 2012;158(2):224. doi: 10.1016/j.jconrel.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solorio LD, Dhami CD, Dang PN, Vieregge EL, Alsberg E. Stem Cells Transl Med. 2012;1(8):632. doi: 10.5966/sctm.2012-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang PN, Dwivedi N, Phillips LM, Yu X, Bowerman C, Solorio LD, Murphy WL, Alsberg E. 2014 Submitted. [Google Scholar]
- 34.Dang PN, Dwivedi N, Yu X, Phillips LM, Bowerman C, Murphy WL, Alsberg E. 2014 Submitted. [Google Scholar]
- 35.Dang PN, Solorio LD, Alsberg E. Tissue Eng. Part A. 2014 doi: 10.1089/ten.tea.2012.0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin L, Chow KL, Leng Y. J Biomed Mater Res A. 2009;89(2):326. doi: 10.1002/jbm.a.31994. [DOI] [PubMed] [Google Scholar]
- 37.Lian JB, McKee MD, Todd AM, Gerstenfeld LC. J Cell Biochem. 1993;52(2):206. doi: 10.1002/jcb.240520212. [DOI] [PubMed] [Google Scholar]
- 38.Hoemann CD, Lafantaisie-Favreau CH, Lascau-Coman V, Chen G, Guzman-Morales J. J Knee Surg. 2012;25(2):85. doi: 10.1055/s-0032-1319782. [DOI] [PubMed] [Google Scholar]
- 39.Yu X, Khalil A, Dang PN, Alsberg E, Murphy WL. Advanced Functional Materials. 2014;24(20):3082. doi: 10.1002/adfm.201302859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Int J Biochem Cell Biol. 2008;40(1):46. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Toh WS, Liu H, Heng BC, Rufaihah AJ, Ye CP, Cao T. Growth Factors. 2005;23(4):313. doi: 10.1080/08977190500252763. [DOI] [PubMed] [Google Scholar]
- 42.Barry F, Boynton RE, Liu B, Murphy JM. Exp Cell Res. 2001;268(2):189. doi: 10.1006/excr.2001.5278. [DOI] [PubMed] [Google Scholar]
- 43.Moioli EK, Hong L, Mao JJ. Wound Repair Regen. 2007;15(3):413. doi: 10.1111/j.1524-475X.2007.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caron MM, Emans PJ, Cremers A, Surtel DA, Coolsen MM, van Rhijn LW, Welting TJ. Osteoarthritis Cartilage. 2013;21(4):604. doi: 10.1016/j.joca.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Gerstenfeld LC, Shapiro FD. J Cell Biochem. 1996;62(1):1. doi: 10.1002/(SICI)1097-4644(199607)62:1%3C1::AID-JCB1%3E3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 46.Wong M, Carter DR. Bone. 2003;33(1):1. doi: 10.1016/s8756-3282(03)00083-8. [DOI] [PubMed] [Google Scholar]
- 47.Lennon DP, Haynesworth SE, Bruder SP, Jaiswal N, Caplan AI. In vitro cell develop biol. 1996;32(10):602. [Google Scholar]
- 48.Haynesworth SE, Goshima J, Goldberg VM, Caplan AI. Bone. 1992;13(1):81. doi: 10.1016/8756-3282(92)90364-3. [DOI] [PubMed] [Google Scholar]
- 49.Solchaga LA, Penick K, Porter JD, Goldberg VM, Caplan AI, Welter JF. J Cell Physiol. 2005;203(2):398. doi: 10.1002/jcp.20238. [DOI] [PubMed] [Google Scholar]
- 50.Tabata Y, Ikada Y, Morimoto K, Katsumata H, Yabuta T, Iwanaga K, Kakemi M. Journal of Bioactive and Compatible Polymers. 1999;14(5):371. [Google Scholar]
- 51.Park H, Temenoff JS, Holland TA, Tabata Y, Mikos AG. Biomaterials. 2005;26(34):7095. doi: 10.1016/j.biomaterials.2005.05.083. [DOI] [PubMed] [Google Scholar]
- 52.Solorio L, Zwolinski C, Lund AW, Farrell MJ, Stegemann JP. J Tissue Eng Regen Med. 2010;4(7):514. doi: 10.1002/term.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farndale RW, Buttle DJ, Barrett AJ. Biochim Biophys Acta. 1986;883(2):173. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]