

Abstract
Introduction:
Glucose-6-phospahte dehydrogenase deficiency (G6PD) is one of the most common inherited disorders affecting around 400 million people worldwide. Molecular analysis of the G6PD gene identified more than 140 distinct mutations, the majority being single base missense mutations. G6PD Mediterranean is the most common variant found in populations of the Mediterranean area.
Aim:
The aim of our study was to perform molecular characterization of G6PD deficiency in families from the Republic of Macedonia and correlate the findings to disease phenotype.
Patients and methods:
Six patients and seven other family members were selected for genetic characterization, the selection procedure involved clinical evaluation and G6PD quantitative testing. All patients were first screened for the Mediterranean mutation, and subsequently for the Seattle mutation. Mutations were detected using PCR amplification and appropriate restriction endonuclease cleavage.
Results:
Four hemizygote and 3 heterozygous carriers for G6PD Mediterranean were detected. All G6PD deficient patients from this group showed clinical picture of hemolysis, and in 66.6% neonatal jaundice was confirmed based on history data. To our knowledge, this is the first study concerned with molecular aspects of the G6PD deficiency in R. Macedonia.
Conclusion:
This study represents a step towards a more comprehensive genetic evaluation in our population and better understanding of the health issues involved.
Keywords: G6PD deficiency, molecular characterization, genotype-phenotype correlation, Republic of Macedonia
1. INTRODUCTION
Glucose-6-phospahte dehydrogenase (G6PD) enzyme catalyses the first step in the hexose monophospahte pathway, thus providing nicotinamide adenine dinucleotide phosphate (NADPH) essential for a number of biosynthetic and detoxifying reactions (1). Deficiency of G6PD is one of the most common inherited disorders affecting more than 400 million people worldwide (1, 2). The human G6PD gene, which contains 13 exons and is over 20 kb long, has been mapped to the Xq28 region (3, 4). Molecular analysis of the G6PD gene identified more than 140 mutations or combination of mutations (5, 6). Almost all G6PD mutations are single base missense mutations spread throughout the coding region of the gene and causing single amino acid substitutions. The absence of large deletions, frameshift mutations and nonsense mutations supports the concept of G6PD expression being crucial for survival (1, 7). Different mutations are found in different ethnic groups. G6PD Mediterranean is the most common variant found in populations of Southern Europe, the Middle East and the Indian subcontinent (1, 8). G6PD Mediterranean is a class 2 variant characterized by a very low enzyme activity (less than 10% of normal) (1, 9). The Mediterranean mutation represents a C→T transition at nucleotide 563, causing the amino acid replacement 188 Ser→Phe and is associated with a spectrum of deficiency manifestations deficiency including: acute acquired hemolytic anemia, favism and pronounced neonatal hyperbilirubinaemia (1, 2, 8, 10). A previous study in southern Croatia showed a prevalence of G6PD deficiency of 0.44% in both sexes and 0.75% in males (11). Molecular characterization of G6PD-deficient patients in the area revealed at least six mutations, among which a new variant named G6PD Split was described (12). There are studies that refer to the G6PD deficit in the Republic of Macedonia. Fraser et al. based on samples from Macedonia and Dalmatia estimated a 1% prevalence of G6PD deficit in former Yugoslavia (13). Andreeva et al. reported on 1-2% prevalence of G6PD deficit in South-Eastern Macedonia. In a subsequent study, a prevalence of 1.02% among children of Macedonian nationality and 6.63% in Romany children on the territory of Skopje has been shown (14). There have been no reports so far on the genetic basis of G6PD deficiency in Macedonia. In concordance with the literature from close countries in the Mediterranean region we could expect highest occurrence of G6PD Mediterranean mutation in the Macedonian cohort of G6PD deficient patients. Here we describe molecular characterization of G6PD deficiency in families from the Republic of Macedonia (RM) with an overview of phenotype expression.
2. METHODS
2.1. Patients
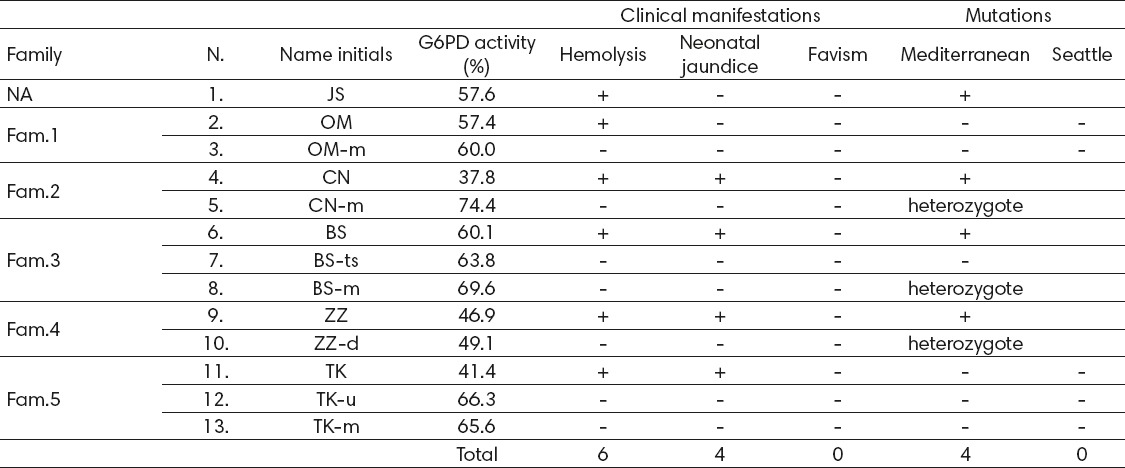
Six patients from the Republic of Macedonia and seven other family members were clinically and genetically evaluated (Table 2). Patients were selected through the Hematology outpatient clinic and Hematology and Neonatology departments. Evaluation for G6PD deficiency was performed in a period not shorter than one month after a hemolytic episode or blood transfusion. Laboratory included: standard blood count and reticulocytes to exclude the possibility of ongoing hemolysis and quantitative testing for G6PD. Peripheral blood anticoagulated with EDTA was obtained for the mutation analysis.
Table 2.
G6PD activity, clinical manifestations and genetic background of the G6PD deficient patients and family members. In the column designated Family, 5 families are represented marked as Fam.1-5. Patient N.1 does not belong to any of the families and was tested individually. Small letters beside the Name initials signify family relation (m-mother, ts- twin sister, d-daughter, and u-uncle). NA – not applicable
The study has been approved by an institutional Ethics Committee in accordance with the Declaration of Helsinki.
2.2. G6PD quantitative assay
The G6PD activity of red blood cells was determined by measuring the rate of increase in nicotinamide adenine dinucleotide phosphate absorbancy at 340 nm using a spectrophotometer (Humananalyser 3000, Germany) and a commercially available kit (AMS U.K. Ltd, East Sussex, U.K.). G6PD activity was calculated in relation to erythrocyte count. Values of 272±27 mU/109 erythrocytes were considered normal. The results were interpreted as a percentage of the normal G6PD activity. Enzyme activity of less than 10% of the normal value was considered a severe deficiency, whereas the activity between 10 and 60% of the normal was considered a moderate deficiency (11, 15).
2.3. Mutation analysis
DNA was extracted from peripheral blood using a standard phenol-chloroform extraction technique. Concentration of DNA was determined spectrophotometrically (Nanodrop 1000, version 3.7.1).
All samples were first examined for the presence of the Mediterranean mutation for which the highest frequency among G6PD deficient patients in this part of the world was expected (1, 2, 8, 10, 12, 16). Subsequently the patients were tested for G6PD Seattle mutation characterized by moderate enzyme deficiency in the context of moderately decreased levels of G6PD activity obtained on the quantitative testing. Further mutation screening was planned for other less common mutations (Cosenza, Union, Cassano, A- etc.) (12, 16, 17, 18).
Detection of G6PD Mediterranean and Seattle mutations was performed using polymerase chain reaction (PCR) amplification and restriction endonuclease cleavage. Synthetic oliconucleotide primer pairs were used for the PCR amplification reaction as previously described (12, 16-19). Amplification was performed using Applied Biosystems 2720 Thermal cycler version 2.08. PCR conditions were: Mediterranean (95°C-10 min, 30 cycles 94°C-45 sec, 58°C-1 min, 72°C-1 min, 72°C-10 min) and Seattle (95°C-10 min, 32 cycles 92°C-2 min, 58°C-1 min, 72°C-2 min, 72°C-10 min).
The 583 bp amplification product of the G6PD Mediterranean polymerase chain reaction was digested with restriction enzyme Mbo II. The Mediterranean mutation creates Mbo II restriction sites in exon 6 that were used to confirm the presence of the mutation. After Mbo II digestion of the PCR amplified product, bands of 379, 120, and 60 bp in normal individuals were obtained, whereas in mutation carriers 4 bands were present (103, 276, 120 and 60 bp) (Table 1) (19).
Table 1.
Detection of G6PD Mediterranean and Seattle mutations using PCR amplification and restriction endonuclease cleavage.
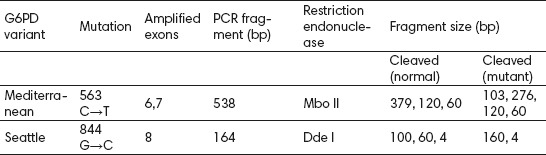
Mutation Seattle in exon 8 was analyzed by Dde I digestion of the 164 bp PCR product. This mutation destroys one restriction site for enzyme Dde I, therefore in mutation carriers 2 fragments would be obtained (160 and 4 bp) instead of 3 (100, 60 and 4 bp) found in normal individuals (Table 1) (12, 16).
Restriction enzyme digestion for the two reactions was performed overnight at 37°C. Digestion products were separated on 3% agarose gel stained with etidium bromide, lighted under UV light and photographed.
3. RESULTS
All results are presented in Table 2.
The first patient (N.1) was tested independently since DNA from other family members could not be obtained during the course of the study. The other 12 patients are grouped in families marked as Fam.1-5.
In three of the patients diagnosis was set prior to this study (N.6 and N.11 were diagnosed by quantitative testing performed in a foreign institution, whereas N.9 was diagnosed by a domestic qualitative assay). This was before the advent of quantitative spectrophotometric testing in our institution. All patients were retested; results of the quantitative testing are presented in Table 2 as percentages of the normal G6PD activity.
All of the 6 G6PD deficiency patients experienced one or more severe hemolytic episodes. When history data were reviewed in detail, in 4 cases neonatal jaundice was confirmed (Table 2). All cases with neonatal jaundice had been treated with phototherapy at maternity wards and followed through neonatology outpatient clinics. In none of the patients exchange transfusion was required. None of the patients showed signs of kernicterus. Favism was not detected in any of the patients.
Patient N.9 was referred to us for consultation on whether his newborn daughter (N.10) should receive preventive parenteral vitamin K that is recognized trigger of hemolysis in G6PD deficient individuals (1, 9, 20).
The results of the genetic testing showed the following: four of the patients were confirmed to be hemizygotes for the Mediterranean mutation (N.1, N.4, N.6 and N.9); also 3 female family members were confirmed heterozygous carriers (mothers of patients N.4 and N.6, and the daughter of patient N.9, marked as N.5, N.8 and N.10 respectively) (Table 2) (Figure 1 and Figure 2). Interestingly, the twin sister of patient N.6 (marked as N.7) was not detected with the Mediterranean mutation (Table 2). This correlates to the normal activity of G6PD obtained by quantitative testing in this patient.
Figure 1.
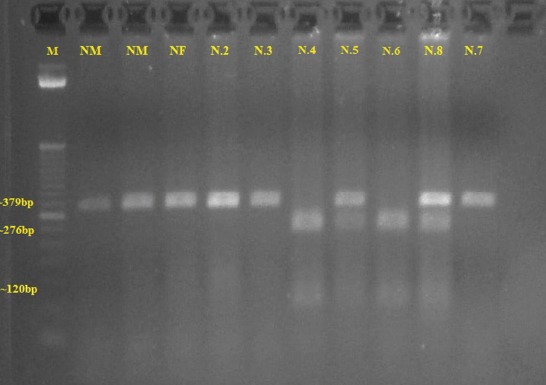
Gel electrophoresis pattern showing DNA fragments for the Mediterranean mutation. Line 1–M, a 50 bp size marker. Lines 2, 3–normal male subjects (NM) Line 4–normal female subject (NF). Lines 5, 6, 11- patients N.2, 3 and 7 negative for the Mediterranean mutation. Lines 7, 9 – patients N. 4 and 6 hemizygotes for the Mediterranean mutation. Lines 8, 10 – patients N.5 and 8 heterozygotes for the Mediterranean mutation
Figure 2.
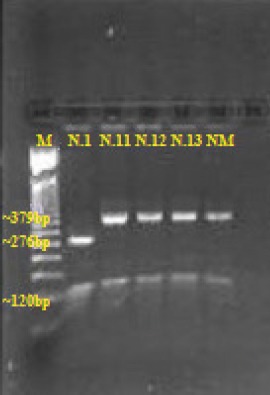
Gel electrophoresis pattern showing DNA fragments for the Mediterranean mutation. Line 1–M, a 50 bp size marker. Line 2- patient N.1, hemizygote for the Mediterranean mutation. Lines 3-5–patients N.11-13, negative for the Mediterranean mutation. Line 6–normal male subject (NM)
None of the examined patients tested positive for the G6PD Seattle mutation (Table 2) (Figure 3).
Figure 3.
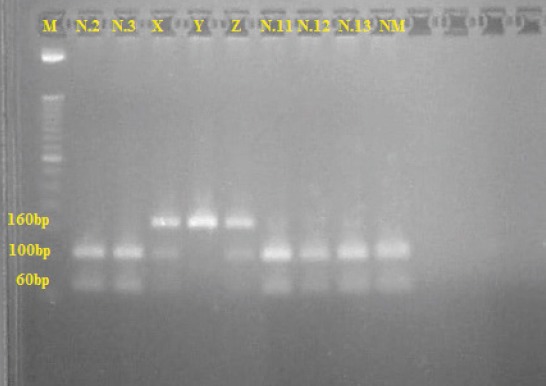
Gel electrophoresis pattern showing DNA fragments for the Seattle mutation. Line 1–M, a 50 bp size marker. Lines 2, 3, 7-9–patients N.2, 3, 11, 12 and 13, negative for the Seattle mutation. Lines 4-6 – family from southern Croatia positive for the Seattle Mutation; lines 4 and 6 (X and Z) female heterozygotes; line 5 (Y) male hemyzigote. Line 10–normal male subject (NM)
4. DISCUSSION
G6PD deficiency has been recognized as one of the most common hereditary enzymopathies affecting hundreds of millions of people worldwide (1, 2, 9). The human G6PD gene comprised of 13 exons has been mapped to Xq28 (3, 4). The majority of G6PD gene mutations affecting the coding sequence are single base missense mutations, also a number of small, in-frame deletions have been found. The lack of large deletions or frameshift mutations supports the vital role of G6PD during human development (1, 2, 5-8). The recent G6PD prevalence estimates in malaria-endemic countries predicted the highest median prevalence of the disease across sub-Saharan Africa and the Arabian Peninsula and lower prevalence in Asia (21). Various reports exist on the prevalence of G6PD deficit in the Mediterranean area. In Italy, a frequency of 0.2-4.4% was reported with the highest frequency of 10% among males in Sardinia (16). In Greece, the prevalence obtained by neonatal screening was 3.14% (22) and in Croatia 0.75% prevalence in males and 0.44 in both sexes was found (11). The highest prevalence was found among the Kurdish Jews of 70% (23). The prevalence in the coastal provinces of Iran was estimated at 8.6-16.4% (24). In Macedonia a prevalence of 1.02% among Macedonians and 6.63% in Romanies was reported (14). There have not been more recent studies to address the issue of G6PD deficiency in our country. On the other hand, Macedonia has been marked for the majority of its territory as a former hiperendemic malaria area (13); the malaria selection pressure could be expected to act on maintaining the frequency of the G6PD deficit similar to the other Mediterranean region countries. Also, no reports exist on the genetic basis of G6PD deficiency in the Republic of Macedonia.
The aim of our study was to perform molecular characterization in a group of G6PD deficiency patients from the RM. Patients were selected through a clinical procedure, after other differential diagnoses of hemolysis and/or neonatal jaundice had been excluded and the deficit was confirmed by a quantitative test. According to the literature from countries in the Mediterranean region, we could expect the highest occurrence of G6PD Mediterranean mutation in the Macedonian cohort of G6PD deficient patients. G6PD Mediterranean has been considered a ubiquitous variant in the Near East, Greece, Croatia, Bulgaria, Italy, Spain and Mauritius (25). It is a class 2 variant characterized with less than 10% of the normal enzyme activity and severe disease manifestations among which severe hemolysis and pronounced neonatal hyperbilirubinemia complicated by kernikterus are major health concerns (1, 2, 8-10). Higher exchange transfusion rates among G6PD deficient neonates have been reported (26). Based on molecular methods, different frequencies of the G6PD Mediterranean mutation were reported among different population varying from 70% in Italy (18), 66.2% and 75.4% in Mazandaran state and coastal provinces of the Caspian Sea of Iran respectively (24, 27), 50% in Greece (10), 16.6% in Croatia (12) and 87.8% among the Kurdish population of Northern Iraq (28). In our cohort, Mediterranean mutation was detected in four deficiency patients and 3 female family members. However, the mutation was not detected in the twin sister of one of the deficiency patients confirming rules of X-linked inheritance in a single pregnancy. Quantitative testing did not show severe deficiency of less than 10% of the normal in any of the G6PD deficiency patients or substantially low levels in the heterozygous carriers of the mutation. We can assume that the reference ranges of the commercially available test were set too high. It would be more appropriate to establish population specific reference ranges and to compare results to those ranges. However rationale of clinical judgment compiled with laboratory results can lead to suspicion of a G6PD deficiency, thus a molecular analysis can be requested. The G6PD Seattle mutation causes G→C transition at nucleotide 844 in exon 8 leading to amino acid replacement at position 285 Arg→His (12, 16). This mutation cases a moderate enzyme deficiency and was not detected in any of our patients. Among our G6PD deficient patients, hemolysis, either drug or infection induced, was the most frequent clinical manifestation present in all cases (100%). Neonatal jaundice on the other hand was present in 4 (66.6%) of the patients. Favism associated with G6PD deficiency has been known since antiquity and has been associated with class 2 variants (1). None of our G6PD deficiency patients manifested favism. Fava beans (Vicia faba) are not a regular component of the Macedonian cuisine. Therefore, cases of favism in RM would most probably be less frequently encountered.
To our knowledge, this is the first study concerned with molecular aspects of the G6PD deficiency in the Republic of Macedonia. The major disadvantage of the study is the small cohort of patients tested. This group of patients by no means represents all the G6PD deficiency patients in the RM. Further more comprehensive studies are warranted. This study is an attempt to start paving the pathway of better understanding this important health issue in our population.
Footnotes
CONFLICT OF INTEREST: NONE DECLARED
REFERENCES
- 1.Beutler E. G6PD deficiency. Blood. 1994;84:3613–3636. [PubMed] [Google Scholar]
- 2.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 3.Martini G, Toniolo D, Vulliamy T, et al. Structural analysis of the X-linked gene encoding human glucose 6-phosphate dehydrogenase. EMBO J. 1986;5:1849–1855. doi: 10.1002/j.1460-2075.1986.tb04436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takizawa T, Huang IY, Ikuta T, Yoshida A. Human glucose-6-phosphate dehydrogenase: primary structure and cDNA cloning. Proc Natl Acad Sci USA. 1986;83:4157–4161. doi: 10.1073/pnas.83.12.4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutler E, Vulliamy TJ. Hematologically important mutations: glucose-6-phosphate dehydrogenase. Blood Cells Mol Dis. 2002;28(2):93–103. doi: 10.1006/bcmd.2002.0490. [DOI] [PubMed] [Google Scholar]
- 6.Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the “old” and update of the new mutations. Blood Cells Mol Dis. 2012;48(3):154–165. doi: 10.1016/j.bcmd.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Ho HY, Cheng ML, Chiu DT. G6PD— anold bottle with new wine. Chang Gung Med J. 2005;28:606–612. [PubMed] [Google Scholar]
- 8.Beutler E. G6PD: population genetics and clinical manifestations. Blood Rev. 1996;10(1):45–52. doi: 10.1016/s0268-960x(96)90019-3. [DOI] [PubMed] [Google Scholar]
- 9.WHO Working Group. Update/Le point. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. 1989;67:601–611. [PMC free article] [PubMed] [Google Scholar]
- 10.Molou E, Schulpis KH, Thodi G, et al. Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency in Greek newborns: the Mediterranean C563T mutation screening. Scand J Clin Lab Invest. 2014;74(3):259–263. doi: 10.3109/00365513.2013.879733. [DOI] [PubMed] [Google Scholar]
- 11.Krzelj V, Zlodre S, Terzic J, Mestrovic M, Jaksic J, Pavlov N. Prevalence of G-6-PD deficiency in the Croatian Adriatic Coast population. Arch Med Res. 2001;32:454–457. doi: 10.1016/s0188-4409(01)00301-0. [DOI] [PubMed] [Google Scholar]
- 12.Barisic M, Korac J, Pavlinac I, et al. Characterization of G6PD deficiency in southern Croatia: description of a new variant, G6PD Split. J Hum Genet. 2005;50:547–549. doi: 10.1007/s10038-005-0292-2. [DOI] [PubMed] [Google Scholar]
- 13.Fraser GR, Grunwald P, Stamatoyannopoulos G. Glucose-6-phosphate dehydrogenase (G6PD) deficiency, abnormal haemoglobins, and thalassaemia in Yugoslavia. J Med Genet. 1966;3:35–41. doi: 10.1136/jmg.3.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreeva M, Efremov G, Markovska P, et al. Urođeni deficit glukoza-6-fosfat dehidrogenaze i hemoglobinopatije na teritoriji Skoplja. Jug pedijat. 1982;25:19–26. [Google Scholar]
- 15.Beutler E, Blume KG, Kaplan JC, Löhr GW, Ramot B, Valentine WN. International Committee for standardization in hematology: recommended methods for red cell enzyme analysis. Br J Haematol. 1977;35:331–340. doi: 10.1111/j.1365-2141.1977.tb00589.x. [DOI] [PubMed] [Google Scholar]
- 16.Calabro V, Mason PJ, Filosa S, et al. Genetic heterogeneity of glucose-6-phosphate dehydrogenase deficiency revealed by Single-strand conformation and sequence analysis. Am J Hum Genet. 1993;52:527–536. [PMC free article] [PubMed] [Google Scholar]
- 17.Alfinito F, Cimmino A, Ferraro F, et al. Molecular characterization of G6PD deficiency in Southern Italy: heterogeneity, correlation genotype-phenotype and description of a new variant (G6PD Neapolis) Br J Haematol. 1997;98(1):41–46. doi: 10.1046/j.1365-2141.1997.1512967.x. [DOI] [PubMed] [Google Scholar]
- 18.Martinez di Montemuros F, Dotti C, Tavazzi D, Fiorelli G, Cappellini MD. Molecular heterogeneity of glucose-6-phosphate dehydrogenase (G6PD) variants in Italy. Haematologica. 1997;82(4):440–445. [PubMed] [Google Scholar]
- 19.Terzić J, Krzelj V, Drmić I, et al. Genetic analysis of the glucose-6-phosphate dehydrogenase deficiency in a southern Croatia. Coll Antropol. 1998;22(2):485–489. [PubMed] [Google Scholar]
- 20.Drugs that should be avoided-official list. G6PD deficiency, Favism association Web site. [Accessed March 22 2014]. http://www.g6pd.org/G6PDDeficiency/SafeUnsafe.aspx.
- 21.Howes RE, Piel FB, Patil AP, et al. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatisticalmodel-based map. PLoS Med. 2012;9(11):e1001339. doi: 10.1371/journal.pmed.1001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Missiou-Tsagariki S. Screening for G-6-PD deficiency as a preventive measure: Prevalence among 1.286.000 Greek newborn infants. J Pediat. 1991;119:293–299. doi: 10.1016/s0022-3476(05)80747-4. [DOI] [PubMed] [Google Scholar]
- 23.Oppenheim A, Jury CL, Rund D, Vulliamy TJ, Luzzatto L. G-6-PD Mediterranean accounts for the high prevalence of G6PD deficiency in Kurdish Jews. Hum Genet. 1993;91:293–294. doi: 10.1007/BF00218277. [DOI] [PubMed] [Google Scholar]
- 24.Mesbah-Namin SA, Sanati MH, Mowjoodi A, Mason PJ, Vulliamy TJ, Noori-Daloii MR. Three major glucose-6-phosphate dehydrogenase-deficient polymorphic variants identified in Mazandaran state of Iran. Br J Haematol. 2002;117(3):763–764. doi: 10.1046/j.1365-2141.2002.03483.x. [DOI] [PubMed] [Google Scholar]
- 25.Kiani F, Schwarzl S, Fischer S, Efferth T. Three-dimensional modeling of glucose-6-phosphate dehydrogenase-deficient variants from German ancestry. PLoS One. 2007;2(7):e625. doi: 10.1371/journal.pone.0000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celik HT, Günbey C, Unal S, Gümrük F, Yurdakök M. Glucose-6-phosphate dehydrogenase deficiency in neonatal hyperbilirubinaemia: Hacettepe experience. J Paediatr Child Health. 2013;49(5):399–402. doi: 10.1111/jpc.12193. [DOI] [PubMed] [Google Scholar]
- 27.Noori-Daloii MR, Hajebrahimi Z, Najafi L, et al. A comprehensive study on the major mutations in glucose-6-phosphate dehydrogenase-deficient polymorphic variants identified in the coastal provinces of Caspian Sea in the north of Iran. Clin Biochem. 2007;40(9-10):699–704. doi: 10.1016/j.clinbiochem.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Al-Allawi N, Eissa AA, Jubrael JM, Jamal SA, Hamamy H. Prevalence and molecular characterization of Glucose-6-Phosphate dehydrogenase deficient variants among the Kurdish population of Northern Iraq. BMC Blood Disord. 2010;10:6. doi: 10.1186/1471-2326-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]